The Biology of Integrins
Adhesion molecules have traditionally been thought of simply as receptors that permit anchorage to other cells or to the underlying extracellular matrix (ECM). However, within the past decade it has become apparent that adhesion molecules such as integrins mediate critical cytosolic signaling events that have a dramatic impact upon cell proliferation, survival, and motility. Integrins act to regulate both physiologic and pathologic events, including complex processes such as angiogenesis, tumor growth, and metastasis. For these reasons, integrins have become attractive targets for drug development, and several effective integrin antagonists are now under clinical evaluation. In turn, the use of integrin-targeted reagents has provided additional mechanistic insights into the workings of the receptor. In particular, it has become apparent that integrins are "mechanosensory" receptors that operate in a context-dependent manner. While integrins that ligate substrate-immobilized ligands typically transduce positive signals into the cell, antagonized or unligated integrins promote negative signaling into the cell, leading to cell cycle arrest or apoptosis. Thus, integrins appear to fulfill a biosensor function, wherein they constantly interrogate the local ECM, and modulate cell behavior accordingly. These new roles that integrins play reinforce the choice of integrins as a therapeutic target, even as they lead us to reassess and optimize current clinical strategies.
ABSTRACT: Adhesion molecules have traditionally been thought of simply as receptors that permit anchorage to other cells or to the underlying extracellular matrix (ECM). However, within the past decade it has become apparent that adhesion molecules such as integrins mediate critical cytosolic signaling events that have a dramatic impact upon cell proliferation, survival, and motility. Integrins act to regulate both physiologic and pathologic events, including complex processes such as angiogenesis, tumor growth, and metastasis. For these reasons, integrins have become attractive targets for drug development, and several effective integrin antagonists are now under clinical evaluation. In turn, the use of integrin-targeted reagents has provided additional mechanistic insights into the workings of the receptor. In particular, it has become apparent that integrins are "mechanosensory" receptors that operate in a context-dependent manner. While integrins that ligate substrate-immobilized ligands typically transduce positive signals into the cell, antagonized or unligated integrins promote negative signaling into the cell, leading to cell cycle arrest or apoptosis. Thus, integrins appear to fulfill a biosensor function, wherein they constantly interrogate the local ECM, and modulate cell behavior accordingly. These new roles that integrins play reinforce the choice of integrins as a therapeutic target, even as they lead us to reassess and optimize current clinical strategies.
The purpose of this treatise is to provide a context for discussion of integrin-targeted therapeutics in cancer. Understanding how integrins function is central to both designing efficient studies that focus on integrins as therapeutic targets and in the interpretation of these and prior studies. Despite great interest and focused efforts, our understanding of these molecules remains incomplete. Indeed, although integrin antagonists were designed 2 decades ago, a number of surprises in the past decade or so have changed how integrins are viewed as therapeutic targets.
The Extracellular Matrix
The extracellular matrix (ECM) is a complex, interacting meshwork of complex glycans and modular glycoproteins that serve numerous physiologic roles. The ECM has historically been referred to simply as the "glue" that holds cells together or as a barrier that defines and maintains the anatomic parameters of tissues. Research over the past 2 decades or so has clearly shown that in addition to obvious structural roles, the ECM plays a critical role in regulating cell behavior during both physiologic and pathologic processes. Cells that bind to the ECM can "sense" mechanical forces that act on the matrix via cell surface receptors, principally integrins.[1] Integrins, in turn, couple to the actin cytoskeleton via a large array of cytosolic signaling and adaptor proteins, many of which were already identified a decade ago.[2-8] This linkage permits cells to translate mechanical stresses into triggers for an array of distinct cell-signaling cascades. Aside from the capacity to interact mechanically with cells, the ECM also acts as a depot for a number of cellular growth factors,[9] many of which require integrin ligation as a cofactor for their signaling activities,[10,11] closely linking the control of cell signaling to the local ECM milieu. These ECM-bound growth factors can be liberated via a number of mechanisms, including the proteolytic "remodeling" of the ECM that occurs during processes such as angiogenesis, wounding, or tumorigenesis.[12,13] The ECM therefore provides a critical scaffold for conferring chemical and mechanical information to the cell.
When considering ECM function as it relates to either the physiology or pathology associated with a tissue or organism, it can be useful to classify the ECM as existing in two different states. Every tissue has an intrinsic anatomic ECM that is present and functions in routine scaffolding, anchorage, and signaling, as described. These matrices are largely composed of collagens, laminins, and proteoglycans, although numerous additional components can be found. However, when tissue remodeling is induced as a pathologic or physiologic process, the preexisting ECM is commonly proteolyzed, and new ECM derived from the plasma or de novo synthesis is deposited. This remodeling ECM is dynamic, and is considered "provisional" due to the rapidity with which it is deposited, assembled, and subsequently proteolyzed.[13-15] The rapid deposition is largely due to the incorporation of plasma proteins such as fibronectin, vitronectin, and fibrinogen, though this ECM also includes many proteins induced de novo among the cells within the affected tissue. Although the original and the provisional ECMs differ in composition, the glycoproteins that compose the ECMs share an important propertyboth sets of ECM are principally bound by integrins.
Integrins
The term "integrins" was initially coined by Hynes to reflect the capacity of these receptors to integrate extracellular and intracellular responses.[16] In particular, the name reflected the capacity of fibronectin-binding integrins to align the actin filament bundles in adherent cells with the alignment of extracellular fibronectin fibrils. Integrins appear to be obligate heterodimers consisting of an α and β subunit that pair in the endoplasmic reticulum prior to transport to the cell surfacealthough anomalies have been reported.[17,18] The two dozen known heterodimers form from the 8 β and the 18 α subunits,[19] with each specific pairing determining the ligand or ligands bound by that heterodimer.
The combined mass of the integrin heterodimer, which can approach ~300 kDa, was observed to be much larger than other known ECM binding molecules, such as the 15 kDa and YGSIR-binding laminin receptors.[20] This suggested functional complexity continues to be described to this day. For example, it has become clear that integrins can be regulated at the conformational level in response to both extracellular and intracellular cues.[21] Despite having no catalytic function and bearing relatively short (30–50 amino acid) cytosolic regions,[16] integrins mediate their own unique form of cell signaling. Signaling occurs, at least in part, via lateral clustering in the cell membrane and the subsequent assembly of multiprotein focal adhesion complexes on the cytosolic face of the membrane.[22]
In addition to lateral associations with other integrins, the integins were found to associate with other cell surface molecules, such as receptor tyrosine kinases. Of particular interest, integrins were found to associate with a number of growth factor receptors and to act as critical coreceptors for growth factor signaling, among these the epidermal growth factor receptors (EGFRs),[23] the platelet-derived growth factor receptors (PDGFRs),[24] and vascular endothelial growth factor receptors (VEGFRs).[10,25]
Cellular Expression of Integrins
A given cell does not express all 24 integrins. Integrins are expressed in a cell-specific manner, and the expression of certain integrins is restricted to cells of a particular lineage. Integrin β2, for example, is expressed on leukocytes.[26] A cell will typically express 8 to 12 distinct hetero-dimers. The regulation of integrin expression is complex (Figure 1), as each subunit is encoded by a different gene, and the expressed proteins then compete for compatible pairs in the endoplasmic reticulum. For example, the β1 integrin can be found on the cell surface paired with any one of 12 different α subunits.[19] Each heterodimer is also subjected to posttranslational processing, which may determine transport to the surface or stability of the heterodimer.


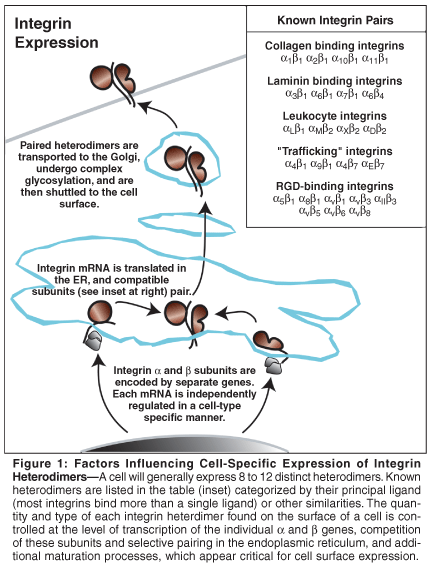
The cellular repertoire of integrins is subject to change in response to specific cues. In the case of the β1 integrins, for example, a relatively low level of expression is observed on the surface of lymphocytes, but this level is dramatically upregulated following T-cell stimulation.[27] This upregulation promotes T-cell interactions with a number of ECM proteins, including fibronectin and collagen. These interactions are physiologically significant, as signaling downstream of integrin ligation lowers the threshold for further stimulation via the antigen receptor complex.[28]
These types of alterations in integrin expression, in which integrin expression is observed to "match" the local ECM, are also common during tissue remodeling. Vascular endothelial cells that are stimulated by angiogenic cytokines express αvβ3 de novo.[29] The expression of this integrin is striking, as integrin αvβ3 is expressed in few cellsonly a limited number of hematopoietic cells in the adult (including osteoclasts). This integrin permits an array of interactions with provisional ECM proteins or protein fragments, permitting cellular interaction with the remodeling ECM and initiating appropriate cellular responses.[12]
These two examples represent changes in integrin expression that are "programmed" into an organism's physiologic response to a given cue or challenge. Both phenomena are conserved across species. The reason for such changes seems self-evident: the quantity and quality of cellular interactions with the ECM will be directly dictated by the repertoire of integrins on the cell surface. These interactions will subsequently influence cellular responses to ECM remodeling and to growth/differentiation factors.
Integrin Ligation and Signaling
The typical ligand of an integrin is a glycoprotein immobilized within the ECM scaffold via polymerization and possible covalent cross-linking to other ECM components. The ligation of integrins by such an immobilized substrate permits cell anchorage, integrin clustering, and engagement of the cell- signaling "machinery." Most of our current understanding of integrin signaling comes from studies in which unpolymerized substrates are immobilized on rigid plastic tissue culture plastic. Though limited in scope, these tissue-culture approaches can give an indication of the types of signaling events that occur in vivo.
Although both α and β integrins have small cytosolic domains, integrin clustering at sites of interaction with the ECM triggers the assembly of a focal adhesion complex containing numerous cytosolic kinases and adaptor proteins that collectively mediate the "downstream signaling" effects of integrins. These include focal adhesion kinase[30] and integrin-linked kinase,[31] small GTPases of the Ras[8] and Rho[32] families, NF-κ[33] and phosphoinositide 3'kinase[34] signaling, as well as activation of a number of different mitogen-activated kinases.[30,35] These signals elicit a range of cellular responses, altering gene transcription while promoting cell survival in the presence of chemical or physical insults, mediating cell migration, and/or inducing cell proliferation. Thus, the integrin interaction with the ECM plays a critical role in modulating cellular responses. However, these signaling events generally require that integrins bind substrate-anchored (ie, immobilized or rigid) ligands, which provide mechanical resistance that permits tensional forces to be generated via the acto-myosin system.[36]
A number of small soluble molecules that have been modeled on ECM have been developed. These include both linear and higher affinity cyclic arginine-glycine-aspartic acid (RGD) peptides[37,38] modeled directly on integrin ligands, as well as small organic mimetics.[39] In contrast to immobilized ligands, these peptides do not elicit cell signaling,[40] or else induce a limited signaling response, as is the case with viral particles.[34,41] Interestingly, if these small soluble ligands are immobilized in a matrix or on a plastic surface, they demonstrate a "rescued" capacity to mediate signaling events. Thus, integrin signaling appears intimately associated with the ability of the ligand to provide mechanical resistance, independent of the ligation event itself.[42] Experiments in which ligated integrins were physically manipulated would appear to support this contention,[43] as would alterations to the local ECM which control the rigidity of the local ECM.[44]
These observations illustrate a duality in integrin function: the same "ligand" can act as a functional agonist or an antagonist, depending upon the contextsoluble, rigid, or flexiblein which it is presented to the cell. The complexity increases when one considers that most ECM proteins are composed of many domains and can have several integrin binding sites, and that these same molecules are polymers/multimers in vivoand moreover that they also bind non-integrin cell surface molecules. The complexity increases a step further when one considers the action of proteases during tissue remodeling. High local concentrations of soluble protein fragments, for example, may compete with native ligand, altering integrin binding to "substrate" ligands and altering cell signaling. Conversely, at low concentrations of soluble ligand, well below those required for blocking adhesion, one may observe an accelerated turnover of the integrin ECM contacts as release is promoted and/or integrins are activated, but the formation of new contacts is not inhibited.[45,46] Such "rebinding" effects can actually promote integrin signaling, and thus provide a concern for consideration in the dosing of small molecules designed to inhibit integrin function.
Integrins and Angiogenesis
Angiogenesis, the growth of new blood vessels from preexisting blood vessels, is stimulated by a number of factors including both proteins and bioactive lipids[10]. Normally quiescent, endothelial cells react to these factors by becoming proliferative and altering protein transcription to effect changes in the expression and function of a number of elements, including integrins. As mentioned above, integrin αvβ3 becomes highly upregulated on endothelial cells during angiogenesis. Soluble antagonists of this integrin can inhibit angiogenesis in vivo,[29,47-49] which, together with the observation that this integrin is expressed in a number of avian and mammalian systems, suggests a central role for this integrin in the regulation of angiogenesis.
Studies have shown that antagonists of integrin αvβ3 can induce endothelial cell apoptosis in vivo, providing a putative mechanism for their antiangiogenic activity.[48,49] The investigations of the Cheresh and Ruoslahti labs revealed that integrins work cooperatively with growth factor receptors such as VEGFRs, PDGFRs, and bFGFRs to mediate downstream signaling via Ras and Raf to MAP kinases.[10,50] Many integrins thus share the capacity to activate the Ras to MAPK pathway either directly or cooperatively with growth factor receptors. Signaling events mediated along this pathway promote survival via both direct and indirect mechanisms. On one hand, kinases within and peripheral to the MAPK pathway can phosphorylate and modify the activity of cytosolic apoptosis-regulating proteins such as bcl-2.[51] On the other hand, alterations in the transcription of prosurvival factors can also change the steady-state levels of survival regulating proteins, such as bax or p53.[52] Altogether, the data suggest that integrins collaborate with growth factors to prevent apoptosis induced by other environmental cues in the local environment.
New Roles for Integrins in Regulating Cell Surival
Despite the discoveries into the mechanisms by which integrins signaled to prevent apoptosis induced by exogenous insults, a growing paradox epitomized in the αvβ3 literature existed. If signaling through an integrin, such as αvβ3, was critical for angiogenesis, why did mice in which the β3 was knocked out have an apparently normal vasculature?[53] Or perhaps more relevant to human disease: why was no vascular defect reported in human patients (a subpopulation of Ashkenazi Jews with Glanzmann thrombasthenia) who lacked functional β3 integrins?[54] Despite a severe bleeding disorder (due to critical function of the integrin on platelets), no vascular disorders were noted.
Finally, although integrins collaborated with specific angiogenic growth factors, most in vivo milieus would provide a number of different growth stimuli, so it was not clear why antagonism of a single class of integrin (eg, αvβ3) on a cell, which expressed 8 to 10 distinct heterodimers, would lead to apoptosis. As mentioned above, the role of this integrin on endothelial cells of a wide range of vertebrates was a reproducible, conserved phenomenon. How could such a conserved phenomenon, apparently critical for survival, be dispensable?
To begin to understand how integrins could influence cell behavior, we initiated in vitro studies with cells engineered to lack or express different levels of integrin αvβ3. Surprisingly, we found that expression of β3 integrin could be selectively harmful (in fact, proapoptotic) to cells within an environment that was deficient in ligands for this integrin.[55] Our studies suggested that the apoptosis that we observed under these circumstances, which we termed "integrin-mediated death" (IMD), was mediated by the cytosolic domain of integrin β3, β1, and possibly other β integrins. The localization of the domain was important, since expression of membrane-associated integrin cytosolic domain fusion proteins resulted in caspase 8–dependent apoptosis, while soluble integrin cytosolic domains did not. Moreover, immobilization of the fusion proteins onto a rigid substrate (by using plastic surfaces coated with antibodies to the extracellular domain of the protein) reconstituted integrin ECM-binding function, engaging the actin cytoskeleton and preventing apoptosis. While the expression of a fusion protein is clearly an artificial system, and was the only circumstance in which IMD was observed in 2D cell culture, it nevertheless served to implicate caspase 8 as a critical effector downstream of unligated or antagonized integrins. Similar dependence has now been described by several other groups.[56-58]
Integrins as Dependence Receptors
These results were interesting in part because caspase 8 is considered to be activated selectively by members of the "death receptor" family. Our data supported the notion that other cellular systems could act to regulate this caspase. In particular, several dependence receptors require the presence of their ligands to maintain cell survival, and some of these, such as APP (Alzheimer's precursor protein), mediate apoptosis via activation of caspase 8.[59] The clustering of APP with integrins in neuronal cells and a shared "NPxY" motif within a small cytosolic domain suggest that integrins may function as dependence receptors, possibly representing a unique subtype.
However, integrins also play a functional role in invasion, mediating anchorage, and downstream signaling to several actin-mobilizing signaling cascades. To begin to understand which role of αvβ3 was critical during angiogenesis, we induced angiogenesis in the chick chorioallantoic membrane with bFGF and treated the embryo intravenously with caspase inhibitors that block IMD in vitro. We reasoned that under these circumstances we would block apoptosis, but if the integrin was required for invasion, then angiogenesis should still be compromised. However, we found that inhibition of caspase 8 selectively rescued angiogenesis in the presence of αvβ3-blocking antibodies (Figure 2), suggesting that the critical role for the integrin was to regulate cell survival. Similar results have been observed in vivo using antagonists of integrin α5β1.[56]


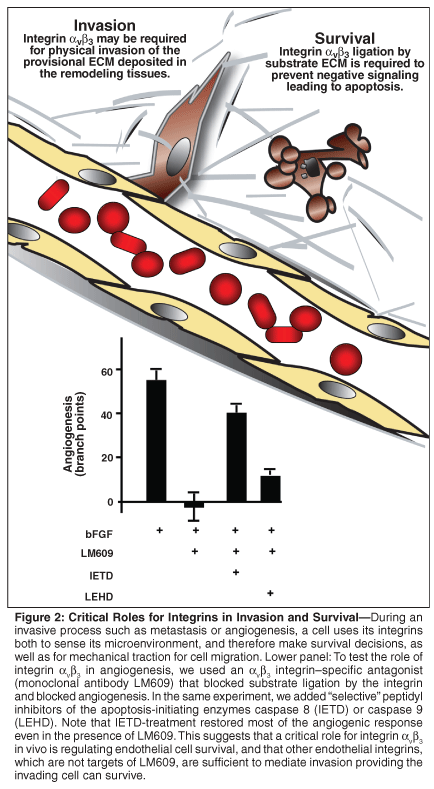
Implications of Positive and Negative Signaling by Integrins
This finding was interesting in many respects, because it suggested that integrins could function both positively and negatively to regulate cell survival. We speculated that integrins may act as biosensors, regulating cell survival in response to cues from the local microenvironment, and in particular the ECM. In the presence of appropriate substrate-immobilized ligands, most integrins appear to transduce positive signals into the cell. In the absence of ligand, or in the presence of soluble antagonists, at least some integrins induce negative signaling, which can lead to apoptosis. From a physiologic standpoint, this model was attractive for a number of reasons. For example, it would help explain how endothelial cells can be cued to undergo apoptosis during the process of "vascular pruning" during the resolution phase of wound healing/tissue differentiation, and it would provide a homeostatic checkpoint that could prevent a cell from migrating out of its appropriate ECM milieu.
Nonetheless, IMD appeared highly complex and dependent upon several factors. The negative signaling was "dose dependent," thus, cells expressing more unligated/antagonized integrins were more susceptible to apoptosis than cells with fewer unligated integrins. Moreover, "crosstalk" with ligated integrins can influence the life or death choice. In particular, IMD does not generally occur on rigid tissue culture plastic, but appears to require a more physiologic matrix, such as a tissue or a three-dimensional gel. Finally, IMD was not a universal phenomenon, as we observed several tumor cell lines which appeared resistant to IMD, presumably due to deficiencies in the expression or regulation of apoptotic proteins necessary for IMD that may have occurred during transformation.[55]
Roles for Integrins in Regulating Metastasis
Based on these observations, we speculated that loss of those apoptotic components that support IMD responses might promote tumor dissemination in vivo. In this respect, caspase 8 expression has been described to be lost or silenced in several metastatic tumors, though it was suggested that the loss of caspase 8 likely contributed to resistance to death receptor–mediated killing. Our studies focused on neuroblastoma (NB), a common solid pediatric tumor in which highly malignant disease (stage IV) exhibits loss of caspase 8 in 70% to 90% of patients.[60] We used several neuroblastoma lines that had been isolated from bone marrow (thus, each had some capacity to metastasize), and examine the ability of these tumors to grow in a developmental tumor modelthe chorioallantois of a chick embryo.[61] We found that 100% of the tumor grew in this environment, with similar tumor sizes regardless of caspase 8 expression. Moreover, the basal rate of apoptosis was similar in these tumors when assessed by TUNEL staining.
In contrast, those NB that lacked expression of caspase 8 exhibited increased NB survival during local tissue invasion and increased metastatic seeding to distant sites. Reconstitution of caspase 8 expression in these cells reduced metastasis to levels comparable to cell lines expressing endogenous caspase 8. Conversely, knockdown of reconstituted or endogenous caspase 8 expression increased metastasis. However, these observations were unlikely to be due to death receptor–mediated killing, due to the resistance of the NB tested to death receptor agonists and due to the xenograft system used.
Although it was difficult to grow the human NB tumors in SCID mice, we were able to observe metastases after ~12 weeks in some animals. From a few of the larger sites we isolated cell lineseach of which demonstrated some degree of suppression of caspase 8 expression.[61,62] These results were consistent with IMD leading to apoptosis. Although none of the NB lines expressed integrin αvβ3, we observed the total elimination of expression of integrin α3β1, a laminin receptor, from these cells.[62] Interestingly, this integrin has previously been implicated as a tumor suppressor,[63] and our results support a role for this integrin as a metastasis suppressor as well.
While it is not yet clear if this integrin mediates IMD in vivo, we found that populations of neuroblastoma lacking α3β1 were partially rescued in their metastatic potential in the chick models even when caspase 8 was expressed.[61] The tumor cells also exhibited increased survival in 3D matrices in vitro that lacked laminin, suggesting that α3β1 may function like αvβ3 and α5β1, inducing caspase 8–dependent apoptosis when unligated or antagonized.
Conclusions and Perspective
While integrins are critical mediators of cell anchorage, it has become apparent that integrins act as more than simple ECM-binding molecules. The array of integrins on the cell surface interacts with the repertoire of ECM and cell surface ligands available, transmitting information back to the cell in a dynamic fashion. Integrins are uniquely poised to act in this capacity, as integrin signaling is dependent upon both chemical and mechanical cues, and is tied into a wide array of signal transduction pathways that control proliferation, survival, and migration. This multirole function of integrins makes them attractive targets for therapy. Integrin antagonists are likely to impact several pathways in any given cell, and therefore contribute to limiting tumor growth and spread through complementary molecular mechanisms.
Disclosures:
The author has no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
1. Ingber D: Integrins as mechanochemical transducers. Curr Opin Cell Biol 3:841-848, 1991.
2. Clark EA, King WG, Brugge JS, et al: Integrin-mediated signals regulated by members of the rho family of GTPases. J Cell Biol 142:573-586, 1998.
3.Klemke RL, Cai S, Giannini AL, et al: Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 137:481-492, 1997.
4. Ezzell RM, Goldmann WH, Wang N, et al: Vinculin promotes cell spreading by mechanically coupling integrins to the cytoskeleton. Exp Cell Res 231:14-26, 1997.
5. Schlaepfer DD, Hunter T: Signal transduction from the extracellular matrixA role for the focal adhesion protein-tyrosine kinase FAK. Cell Struct Funct 21:445-450, 1996.
6. Parsons JT: Integrin-mediated signalling: Regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol 8:146-152, 1996.
7. McNamee HP, Liley HG, Ingber DE: Integrin-dependent control of inositol lipid synthesis in vascular endothelial cells and smooth muscle cells. Exp Cell Res 224:116-122, 1996.
8. Clark EA, Hynes RO: Ras activation is necessary for integrin-mediated activation of extracellular signal-regulated kinase 2 and cytosolic phospholipase A2 but not for cytoskeletal organization. J Biol Chem 271:14814-14818, 1996.
9. Schonherr E, Hausser HJ: Extracellular matrix and cytokines: A functional unit. Dev Immunol 7:89-101, 2000.
10. Eliceiri BP, Cheresh DA: The role of alphav integrins during angiogenesis. Mol Med 4:741-750, 1998.
11. Giancotti FG, Ruoslahti E: Integrin signaling. Science 285:1028-1032, 1999.
12. Stupack DG, Cheresh DA: Integrins and angiogenesis. Curr Top Dev Biol 64:207-238, 2004.
13. DeClerck YA, Mercurio AM, Stack MS, et al: Proteases, extracellular matrix, and cancer: A workshop of the path B study section. Am J Pathol 164:1131-1139, 2004.
14. Clark RA: Fibronectin matrix deposition and fibronectin receptor expression in healing and normal skin. J Invest Dermatol 94:128S-134S, 1990.
15. Ravanti L, Kahari VM: Matrix metalloproteinases in wound repair (review). Int J Mol Med 6:391-407, 2000.
16. Hynes RO: Integrins: A family of cell surface receptors. Cell 48:549-554, 1987.
17. Meng X, Cheng K, Krohkin O, et al: Evidence for the presence of a low-mass beta1 integrin on the cell surface. J Cell Sci 118:4009-4016, 2005.
18. Coppolino M, Migliorini M, Argraves WS, et al: Identification of a novel form of the alpha 3 integrin subunit: Covalent association with transferrin receptor. Biochem J 306(pt 1):129-134, 1995.
19. Hynes RO: Integrins: Bidirectional, allosteric signaling machines. Cell 110:673-687, 2002.
20. Edgar D: Neuronal laminin receptors. Trends Neurosci 12:248-251, 1989.
21. Ginsberg MH, Partridge A, Shattil SJ: Integrin regulation. Curr Opin Cell Biol 17:509-516, 2005.
22. Shattil SJ, Newman PJ: Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood 104:1606-1615, 2004.
23. Moro L, Dolce L, Cabodi S, et al: Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem 277:9405-9414, 2002.
24. Schneller M, Vuori K, Ruoslahti E: Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J 16:5600-5607, 1997.
25. Borges E, Jan Y, Ruoslahti E: Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J Biol Chem 275:39867-39873, 2000.
26. Larson RS, Springer TA: Structure and function of leukocyte integrins. Immunol Rev 114:181-217, 1990.
27. Springer TA: Adhesion receptors of the immune system. Nature 346:425-434, 1990.
28. Shimizu Y, van Seventer GA, Horgan KJ, et al: Costimulation of proliferative responses of resting CD4+ T cells by the interaction of VLA-4 and VLA-5 with fibronectin or VLA-6 with laminin. J Immunol 145:59-67, 1990.
29. Brooks PC, Clark RA, Cheresh DA: Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264:569-571, 1994.
30. Schlaepfer DD, Hunter T: Integrin signalling and tyrosine phosphorylation: Just the FAKs? Trends Cell Biol 8:151-157, 1998.
31. Dedhar S, Williams B, Hannigan G: Integrin-linked kinase (ILK): A regulator of integrin and growth-factor signalling. Trends Cell Biol 9:319-323, 1999.
32. Juliano RL, Reddig P, Alahari S, et al: Integrin regulation of cell signalling and motility. Biochem Soc Trans 32:443-446, 2004.
33. Courter DL, Lomas L, Scatena M, et al: Src kinase activity is required for integrin alphaVbeta3-mediated activation of nuclear factor-kappaB. J Biol Chem 280:12145-12151, 2005
34. Li E, Stupack D, Klemke R, et al: Adenovirus endocytosis via alpha(v) integrins requires phosphoinositide-3-OH kinase. J Virol 72:2055-2061, 1998.
35. Dolfi F, Garcia-Guzman M, Ojaniemi M, et al: The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc Natl Acad Sci U S A 95:15394-15399, 1998.
36. Schwartz MA, Ingber DE: Integrating with integrins. Mol Biol Cell 5:389-393, 1994.
37. Engleman VW, Nickols GA, Ross FP, et al: A peptidomimetic antagonist of the alpha(v)beta3 integrin inhibits bone resorption in vitro and prevents osteoporosis in vivo. J Clin Invest 99:2284-2292, 1997.
38. Ruoslahti E: RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12:697-715, 1996.
39. Andronati SA, Karaseva TL, Krysko AA: PeptidomimeticsAntagonists of the fibrinogen receptors: Molecular design, structures, properties and therapeutic applications. Curr Med Chem 11:1183-1211, 2004.
40. Schwartz MA: Spreading of human endothelial cells on fibronectin or vitronectin triggers elevation of intracellular free calcium. J Cell Biol 120:1003-1010, 1993.
41. Villaverde A, Feliu JX, Harbottle RP, et al: A recombinant, arginine-glycine-aspartic acid (RGD) motif from foot-and-mouth disease virus binds mammalian cells through vitronectin and, to a lower extent, fibronectin receptors. Gene 180:101-106, 1996.
42. Alenghat FJ, Ingber DE: Mechano-transduction: All signals point to cytoskeleton, matrix, and integrins. Sci STKE 2002(119):PE6, 2002.
43. Chen CS, Mrksich M, Huang S, et al: Geometric control of cell life and death. Science 276:1425-1428, 1997.
44. Giannone G, Sheetz MP: Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol 16:213-223, 2006.
45. Bassler N, Loeffler C, Mangin P, et al: A mechanistic model for paradoxical platelet activation by ligand-mimetic {alpha}IIb{beta}3 (GPIIb/IIIa) antagonists. Arterioscler Thromb Vasc Biol 27(3):e9-e15, 2007 [epub Dec 14, 2006].
46. Legler DF, Wiedle G, Ross FP, et al: Superactivation of integrin alphavbeta3 by low antagonist concentrations. J Cell Sci 114:1545-1553, 2001.
47. Friedlander M, Brooks PC, Shaffer RW, et al: Definition of two angiogenic pathways by distinct alpha v integrins. Science 270:1500-1502, 1995.
48. Brooks PC, Montgomery AM, Rosenfeld M, et al: Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 79:1157-1164, 1994.
49. Storgard CM, Stupack DG, Jonczyk A, et al: Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J Clin Invest 103:47-54, 1999.
50. Hood JD, Frausto R, Kiosses WB, et al: Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J Cell Biol 162:933-943, 2003.
51. Breitschopf K, Haendeler J, Malchow P, et al: Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol 20:1886-1896, 2000.
52. Stromblad S, Becker JC, Yebra M, et al: Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J Clin Invest 98:426-433, 1996.
53. Hodivala-Dilke KM, McHugh KP, Tsakiris DA, et al: Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103:229-238, 1999.
54. Newman PJ, Seligsohn U, Lyman S, et al: The molecular genetic basis of Glanzmann thrombasthenia in the Iraqi-Jewish and Arab populations in Israel. Proc Natl Acad Sci U S A 88:3160-3164, 1991.
55. Stupack DG, Puente XS, Boutsaboualoy S, et al: Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol 155:459-470, 2001.
56. Kim S, Bakre M, Yin H, et al: Inhibition of endothelial cell survival and angiogenesis by protein kinase A. J Clin Invest 110:933-941, 2002.
57. Zhao H, Ross FP, Teitelbaum SL: Unoccupied alpha(v)beta3 integrin regulates osteoclast apoptosis by transmitting a positive death signal. Mol Endocrinol 19:771-780, 2005.
58. Marconi A, Atzei P, Panza C, et al: FLICE/caspase-8 activation triggers anoikis induced by beta1-integrin blockade in human keratinocytes. J Cell Sci 117:5815-5823, 2004.
59. Bredesen DE, Mehlen P, Rabizadeh S: Receptors that mediate cellular dependence. Cell Death Differ 12:1031-1043, 2005.
60. Teitz T, Wei T, Valentine MB, et al: Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med 6:529-535, 2000.
61. Stupack DG, Teitz T, Potter MD, et al: Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 439:95-99, 2006.
62. Teitz T, Stupack DG, Lahti JM: Halting neuroblastoma metastasis by controlling integrin-mediated death. Cell Cycle 5:681-685, 2006.
63. Owens DM, Watt FM: Influence of beta1 integrins on epidermal squamous cell carcinoma formation in a transgenic mouse model: Alpha3beta1, but not alpha2beta1, suppresses malignant conversion. Cancer Res 61:5248-5254, 2001.


MammoScreen AI Tool Improves Diagnostic Performance of Radiologists in Detecting Breast Cancer
December 16th 2020This study demonstrated that the concurrent use of this new artificial intelligence tool alongside mammography improved the diagnostic performance of radiologists in the detection of breast cancer without prolonging their workflow.

Preliminary Results Suggest AB-MR Detects Breast Cancers that 3-D Mammograms May Miss
November 29th 2020This study found that abbreviated breast magnetic resonance imaging as a supplemental screening test in women with dense breasts shows an increase in cancer detection over digital breast tomosynthesis screening.




DeepSurv Displays Possible Benefits in Prognostic Evaluation, Treatment Recommendation
July 20th 2020The deep learning survival neural network model demonstrated the potential to provide personalized treatment recommendations based on real clinical data in patients with non-small cell lung cancer.

