Cancer Management Chapter 27: Non-Hodgkin lymphoma
The incidence rates of non-Hodgkin lymphoma (NHL) in the United States have almost doubled between 1970 and 1990, representing one of the largest increases of any cancer. Although the overall incidence rates of NHL began to stabilize in the late 1990s, the temporal trends varied by histologic subtype. Some of this increase may be artifactual, resulting from improved diagnostic techniques and access to medical care, or directly related to the development of NHL in 25- to 54-year-old men with human immunodeficiency virus (HIV) infection. However, additional factors must be responsible for this unexpected increase in frequency of NHL that has been observed throughout the United States.
The incidence rates of non-Hodgkin lymphoma (NHL) in the United States have almost doubled between 1970 and 1990, representing one of the largest increases of any cancer. Although the overall incidence rates of NHL began to stabilize in the late 1990s, the temporal trends varied by histologic subtype. Some of this increase may be artifactual, resulting from improved diagnostic techniques and access to medical care, or directly related to the development of NHL in 25- to 54-year-old men with human immunodeficiency virus (HIV) infection. However, additional factors must be responsible for this unexpected increase in frequency of NHL that has been observed throughout the United States.
The incidence of NHL per 100,000 persons rose from 8.8 in 1972− 1974 to 19.5 in 2002− 2006 (all histologic subtypes) in the United States. The increases have been more pronounced in whites, males, the elderly, and those with NHL diagnosed at extranodal sites. Similar findings have been reported in other developed countries. NHL rates have decreased among American males aged 25 to 54 years in the middle to late 1990s (6% to 16% per year), although this may be in part due to improved HIV treatment.
Currently, NHL represents approximately 4.6% of all cancer diagnoses (4.8% in males and 4.4% in females), being the fifth most common cancer in women and the sixth in men. Estimates from the American Cancer Society indicate that in 2009, some 65,980 new cases of NHL will be diagnosed in the United States and approximately 19,500 people will die of this disease.
Epidemiology
Gender The overall incidence of lymphoma is higher in men than in women. The incidence rate (per 100,000 population) between 2002 and 2006 was 40% higher in males than in females. Only thyroid NHL is more common in women than in men.
Age Overall, the incidence of NHL rises exponentially with increasing age. In persons older than age 65, the incidence is 87.2 per 100,000 population. Except for high-grade lymphoblastic and Burkitt lymphomas (the most common types of NHL seen in children and young adults), the median age at presentation for all subtypes of NHL exceeds 50+ years. Low-grade lymphomas account for 37% of NHLs in patients between the ages of 35 and 64 years at diagnosis but for only 16% of cases in those younger than age 35.
Race The incidence varies by race, with whites at higher risk than blacks and Asian-Americans (incidence rates increased 40% to 70% in whites compared with blacks). Most histologies, particularly low-grade small lymphocytic and follicular lymphomas, are more common in whites than in blacks. Only peripheral T-cell lymphoma, mycosis fungoides, and Szary syndrome are more common in blacks than in whites.
The InterLymph Consortium recently found that a wide range of putative environmental and genetic risk factors were associated with the risk of developing NHL. They noted commonality and heterogeneity of risk factors (including autoimmune conditions, clinical features, and genetic factors) that were dependent on particular NHL histologic subtypes
(Morton LM et al: Blood 112:5150–5160, 2008)
.
Geography NHL is most common in developed countries, with the United States having the highest rate worldwide. The lowest NHL rates are found in Eastern and south central Asia (2 to 3 per 100,000 population). Certain endemic geographic factors appear to influence the development of NHL in specific areas.
Human T-cell lymphotrophic virus-1 (HTLV-1)-associated adult T-cell lymphoma/leukemia (ATLL) occurs more frequently where HTLV-1 is endemic, in southern Japan and the Caribbean, and occurs sporadically in Brazil, sub-Saharan Africa, the Middle East, and the southeastern United States. The seroprevalence in southwest Japan is 16%, although the lifetime risk of ATLL for these persons is 2% to 6%.
The incidence (per 100,000 population) of Burkitt NHL in Africa (Nigeria and Tanzania) is 6 to 8, as compared with 0.1 in the United States. The clinical features of Burkitt lymphoma in Africa differ from those of cases reported to the American Burkitt Lymphoma Registry. Etiologic endemic factors include malaria as a source of chronic B-cell antigenic stimulation and Epstein-Barr virus (EBV)-induced immortalization of B lymphocytes.
Heavy-chain disease is a disorder of B-lymphoid cells characterized by diffuse thickening of the small intestine due to a lymphoplasmacytic infiltrate with secretion of incomplete IgA heavy chains. Pathologically, it is a mucosa-associated lymphoid tissue (MALT) lymphoma of the small bowel. This clinicopathologic entity is rarely encountered in individuals other than those of Mediterranean ethnic origin.
Follicular lymphomas are more common in North America and Europe but are rare in the Caribbean, Africa, China, Japan, the Middle East, and Latin America.
Peripheral T-cell lymphomas are more common in Europe and China than in North America. They represent 7% to 12% of lymphomas in Western countries.
Disease site The NHLs are a heterogeneous group of neoplasms that usually arise or present in lymphoid tissues, such as lymph nodes, spleen, and bone marrow, but they may arise in almost any tissue. The most frequent sites for extranodal lymphomas, which constitute about 20% to 30% of all lymphomas (peripheral T-cell NHL 80%; extranodal, follicular 9%), are the stomach, skin, oral cavity and pharynx, small intestine, and central nervous system (CNS). Although primary CNS lymphomas are rare, there has been a threefold increase in incidence, even if patients with HIV infection and other types of immunosuppression are excluded.
Survival The 5-year relative survival rate of patients with NHL increased from 28% between 1950 and 1954 to 63% between 1990 and 2003. These improvements in survival occurred mainly in patients with intermediate-high-grade histologies. The potential for cure varies among the different histologic subtypes and is related in part to stage at presentation and response to initial therapy. The natural history (survival rates) for indolent lymphomas has been unchanged from the 1950s to the early 1990s, although recent data including an analysis from Iowa of Surveillance, Epidemiology, and End Results (SEER) data (1979− 1999) shows improving survival rates of patients with follicular lymphoma.
Etiology and risk factors
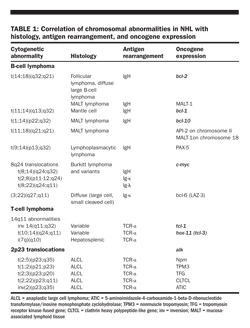
Chromosomal translocations and molecular rearrangements Nonrandom chromosomal and molecular rearrangements play an important role in the pathogenesis of many lymphomas and correlate with histology and immunophenotype (Table 1). The most commonly associated chromosomal abnormality in NHL is the t(14;18)(q32;q21) translocation, which is found in 85% of follicular lymphomas and 28% of higher grade NHLs. This translocation results in the juxtaposition of the bcl-2 apoptotic inhibitor “oncogene” at chromosome band 18q21 to the heavy-chain region of the immunoglobulin locus within chromosome band 14q32.
The t(11;14)(q13;q32) translocation results in overexpression of bcl-1 (cyclin D1/PRAD 1), a cell-cycle− control gene on chromosome 11q13, and is characteristically associated with mantle cell lymphoma. The t(3;16)(q27;p11) translocation makes the gene for the interleukin-2 (IL-2) receptor a partner of bcl-6, which is expressed in diffuse large cell lymphoma.
Chromosomal translocations involving 8q24 lead to c-myc deregulation and are frequently seen in Burkitt and Burkitt-like lymphomas, including those associated with HIV infection.
Environmental factors also may play a role in the development of NHL.
Certain workers have a slightly increased risk of developing NHL, including farmers; pesticide applicators; grain (flour) millers; meat workers; wood and forestry workers; chemists; painters; mechanics; machinists; and workers in the petroleum, rubber, plastics, and synthetics industries.


Chemicals that have been linked to the development of NHL include a variety of pesticides and herbicides (2,4-D-organophosphates, chlorophenols), solvents and organic chemicals (benzene, carbon tetra-chloride), and wood preservatives. There is some evidence that the association between pesticides and NHL risk was limited to t(14;18)-positive NHL cases.
Viruses Several viruses have been implicated in the pathogenesis of NHL, including EBV; HTLV-1; Kaposi-s sarcoma-associated herpesvirus (KSHV, also known as human herpesvirus 8, or HHV-8); and hepatitis C virus (HCV). Meta-analyses have shown 13% to 15% HCV seroprevalence in certain geographic regions among persons with B-cell NHL, especially marginal zone NHL.
EBV is a DNA virus that has been associated with Burkitt lymphoma, particularly in endemic areas of Africa; Hodgkin lymphoma; lymphomas in im-munocompromised patients (ie, organ transplantation and HIV infection); sinonasal lymphoma (Asia and South America); and sporadically in other B- and T-cell lymphomas. In contrast to studies performed in European patients, Mexican patients with intestinal lymphomas show a high frequency of EBV positivity; this finding is not limited to T-cell NHLs but rather includes a significant portion of B-cell NHLs. EBV can transform lymphocytes in culture. B lymphocytes from normal EBV-positive subjects have been shown to grow as tumors in mice with severe combined immunodeficiency.
HTLV-1 is a human retrovirus that establishes a latent infection via reverse transcription in activated T-helper cells. A minority (5%) of carriers develop ATLL. An HTLV-1− like provirus has been detected in some patients with mycosis fungoides, although conflicting findings have been reported.
KSHV-like DNA sequences are frequently detected in primary effusion lymphomas in patients with HIV infection and in those with multicentric (plasma cell variant) Castleman-s disease.
HCV infection is associated with the development of clonal B-cell expansions and certain subtypes of NHL, particularly in the setting of essential (type II) mixed cryoglobulinemia. HCV may predispose B cells to malignant transformation by enhancing signal transduction upon binding to the CD81 (TAPA-1) molecule.
Bacterial infections Infection with Borrelia burgdorferi, the etiologic agent in Lyme disease, has been detected in about 35% of patients with primary cutaneous B-cell lymphoma in Scotland. A near-complete clinical and histologic remission of a primary marginal zone B-cell lymphoma was observed after eradication of B burgdorferi with antibiotic treatment. Gastric MALT lymphoma is seen most frequently, but not exclusively, in association with Helicobacter pylori infection. Recent studies indicate that Campylobacter jejuni and immunoproliferative small intestinal disease (α-chain disease) are related.
Some reports noted an association between infection with Chlamydia psittaci and ocular adnexal lymphoma. The infection was found to be highly specific and does not reflect a subclinical infection widespread among the general population. Responses to antibiotics have been reported. Attempts to confirm this association in the Western hemisphere have been unsuccessful.
Patel and colleagues recently compared the cancer incidence among 54,780 HIV-infected persons (157,819 person-years of follow-up) with the incidence in the general population from 1992 to 2003. Coinciding with increasing use of highly active antiretroviral therapy, the relative incidence of NHL among HIV-infected persons decreased over time compared with the general population [1992–1995: 79.4 (72.4–87.1); 1996–1999: 40.3 (35.6–45.6); 2000–2003: 17.0 (14.3–20.3)]
(Patel P et al: Ann Intern Med 148:728–736, 2008)
.
Immune modulation Congenital and acquired states of immunosuppression that are at increased risk include ataxia-telangiectasia, Wiskott-Aldrich syndrome, common variable hypogamma-globulinemia, X-linked lymphoproliferative syndrome, and severe combined immunodeficiency.
Acquired immunodeficiency states such as HIV infection (relative risk of NHL is 75-100) increased among patients with AIDS compared with the general population (although recent data in the post-HAART [highly active antiretroviral therapy] era suggest it has decreased), usually high grade and extranodal and iatrogenic immunosuppression (ie, organ or blood stem-cell transplantation recipients, long-term survivors of Hodgkin) contribute to an increased risk of NHL.
An increased incidence of GI lymphomas is seen in patients with celiac (nontropical) sprue and inflammatory bowel disease, particularly Crohn-s disease. An aberrant clonal intraepithelial T-cell population can be found in up to 75% of patients with refractory celiac sprue prior to the development of overt T-cell lymphoma using immunophenotyping and T-cell receptor gamma gene rearrangement PCR (polymerase chain reaction) techniques. Systemic lupus erythematosus and rheumatoid arthritis have been associated with B-cell lymphoma. Sjgren-s syndrome has been associated with NHL overall, diffuse large B-cell lymphoma (DLBCL), and marginal zone lymphoma.
Patients who receive chemotherapy and/or radiation therapy are also at increased risk of developing NHL.
Lifestyle factors Several studies have reported an excess risk of NHL in association with diets high in fat and meat products. Some studies have suggested that ultraviolet radiation exposure and alcohol intake may be linked inversely with NHL risk.
Genetic susceptibility Several reports have implicated a role for genetic variants in the risk of NHL, including genes that influence DNA integrity and methylation; genes that alter B-cell survival and growth; and genes that involve innate immunity, oxidative stress, and xenobiotic metabolism. Of note, replication studies are needed to rule out false-positive associations.
Signs and symptoms
Fever, weight loss, and night sweats, referred to as systemic B symptoms, as well as fatigue and weakness, are more common in advanced or aggressive NHL but may be present in all stages and histologic subtypes.
Low-grade lymphomas Painless, slowly progressive peripheral adenopathy is the most common clinical presentation in patients with low-grade lymphomas. Patients sometimes report a history of waxing and waning adenopathy before seeking medical attention. Spontaneous regression of enlarged lymph nodes can occur and can cause a low-grade lymphoma to be confused with an infectious condition.
Primary extranodal involvement and B symptoms are uncommon at presentation; however, both are common in advanced or end-stage disease. Bone marrow is frequently involved, sometimes in association with cytopenias. Splenomegaly is seen in about 40% of patients, but the spleen is rarely the only involved site at presentation.
Intermediate-and high-grade lymphomas The clinical presentation of intermediate- and high-grade lymphomas is more varied. Although the majority of patients present with adenopathy, more than one-third present with extranodal involvement, the most common sites being the GI tract (including Waldeyer-s ring), skin, bone marrow, sinuses, genitourinary (GU) tract, thyroid, and CNS. B symptoms are more common, occurring in about 30% to 40% of patients.
Lymphoblastic lymphoma often presents with an anterior superior mediastinal mass, superior vena cava syndrome, and leptomeningeal disease with cranial nerve palsies.
American patients with Burkitt lymphoma often present with a large abdominal mass and symptoms of bowel obstruction.
Screening and diagnosis
No effective methods are available for screening or identifying populations at high risk of developing NHL. A definitive diagnosis can be made only by biopsy of pathologic lymph nodes or tumor tissue. It is critical to perform an excisional lymph node biopsy (fine-needle aspiration [FNA] is insufficient for diagnostic purposes) to avoid false-negative results and inaccurate histologic classification. One study showed that when FNAs and biopsy results for the diagnosis of NHL and Hodgkin lymphoma were compared, only 12% of FNAs correlated with subsequent excisional biopsy results. When clinical circumstances make surgical biopsy of involved lymph nodes or extranodal sites prohibitive, a core biopsy obtained under CT or ultrasonographic guidance may suffice but often requires the integration of histologic examination for diagnosis. A formal review by an expert hematopathologist is mandatory. Additional studies, such as immunophenotyping and genotyping, are often necessary.
Initial diagnostic evaluation of patients with lymphoproliferative malignancy should include the following:
• Careful history (night sweats, weight loss, fever; neurologic, musculoskeletal, or GI symptoms)
• Physical examination (lymph nodes, including submental, infraclavicular, epitrochlear, iliac, femoral, and popliteal nodes; pericardial rub, pleural effusion, distended neck and/or upper extremity veins in superior vena cava syndrome; breast masses; hepatosplenomegaly, bowel obstruction, renal mass, and testicular or ovarian mass; focal neurologic signs, such as plexopathy, spinal cord compression, nerve root infiltration, and meningeal involvement; skin lesions)
• Biopsy of peripheral lymphadenopathy (excisional biopsy)
• Chest x-ray for evaluation of mediastinal mass
• CT scan of the neck (cervical lymph nodes, Waldeyer-s ring) and chest (mediastinal, hilar, or parenchymal pulmonary disease)
• CT scan of the abdomen and pelvis (enlarged lymph nodes, splenomegaly, filling defects in liver and spleen)
• Bilateral bone marrow biopsy and aspirate
• PET scans (selected cases: aggressive histologies) for staging at diagnosis, response, assessment, and relapse
• CBC with differential and platelet count (peripheral blood lymphocytosis with circulating malignant


cells is common in low-grade and mantle cell lymphomas). Bone marrow and peripheral blood involvement may be present, and the distinction between leukemia and lymphoma is difficult to make in some cases.
• General chemistry panel (including lactic dehydrogenase [LDH] level determination) is mandatory.
• Hepatitis B virus (HBV) and HCV panels should be considered, especially in patients anticipated to receive monoclonal antibody therapy and/or chemotherapy (HBV:HBV surface antigen and HBV core antibody; HCV:HCV antibody).
• HIV serology in at-risk patients with diffuse large cell and other aggressive and Burkitt histologies; HTLV-1 serology in select patients with cutaneous T-cell lymphoma, especially if they have hypercalcemia
• Cytogenetic and molecular analyses of lymph node, bone marrow, and peripheral blood (selected cases)


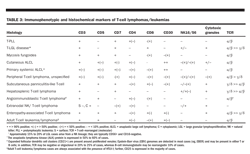
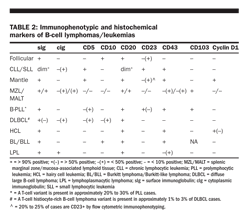
• Immunophenotyping can be of particular benefit in distinguishing B-cell chronic leukemia/small lymphocytic lymphoma from other lymphomas (Table 2). Immunophenotyping and histochemical markers may also be of benefit in distinguishing T-cell lymphomas/leukemias (Table 3).
• Examination of CSF and consideration of intrathecal chemotherapy prophylaxis in patients with (1) diffuse aggressive NHL with bone marrow, epidural, testicular, paranasal sinus, breast, or multiple extranodal sites; (2) high-grade lymphoblastic lymphoma and Burkitt lymphoma and its variants; (3) primary CNS lymphoma if no evidence of increased intracranial pressure
• Upper GI endoscopy and/or GI series with small bowel follow-through in patients with head and neck involvement (tonsil, base of tongue, nasopharynx) and those with a GI primary; mantle cell lymphoma is associated with a high incidence of occult GI involvement.
• Ultrasonography of opposite testis in patients with a testicular primary
• Spinal MRI scan for epidural disease when clinically indicated (useful in the evaluation of suspected spinal cord involvement).
Pathology
The Working Formulation was proposed in 1982 as a modification of the Rappaport classification of NHL based on morphology and biologic aggressiveness. Although many subtypes were not recognized, including T-cell lineage lymphomas, a revised European-American classification of lymphoid neoplasms (REAL classification) was introduced in 1994 incorporating T-cell malignancies, subtypes of Hodgkin lymphoma, and newer defined lymphoma-proliferative disorders. The WHO (World Health Organization) classification for lymphomas (introduced in 1999) uses the principles of



the REAL classification and defines each entity according to morphologic features, immunophenotype, genetic features, postulated normal counterpart, and clinical features. The WHO classification is similar to the REAL classification, with some modifications based on current data (Table 4).
WHO Classification
The most frequently occurring clinical entities recognized by the REAL/WHO classification are DLBCL (31%), follicular lymphoma (22%), lymphoma of MALT type (8%), small lymphocytic lymphoma (7%), mantle cell lymphoma (6%), peripheral T-cell lymphoma (7%), primary mediastinal large B-cell lymphoma (2%), anaplastic large T-/null-cell lymphoma (2%), lymphoblastic lymphoma of T- or B-cell lineage, marginal zone (monocytoid) B-cell lymphoma (2%), lymphoplasmacytic lymphoma (2%), Burkitt lymphoma (2%), and other (9%).
The WHO classification includes three types of follicular lymphoma (grades 1− 3). Grades 1/2 correspond to follicular small-cleaved cell and follicular mixed small-cleaved and large cell lymphoma, which are considered to be low-grade lymphomas by the Working Formulation. Grades 3a and 3b correspond to follicular large cell lymphoma, which is considered an intermediate-grade NHL in the Working Formulation, and is generally treated as a large cell lymphoma (especially grade 3b). The WHO classification considers B-cell small lymphocytic lymphoma to be synonymous with chronic lymphocytic leukemia (CLL).
The classic immunophenotype for MCL is CD5+/CD10–/CD20+/FMC7+/CD23– with bright expression of surface immunoglobulin and CD79b. 53 consecutive patients with untreated MCL had CD23 expression assessed by flow cytometric immunophenotyping (FCIP).By FCIP, 26% of MCL patients had CD23+ disease. Further, long-term event-free survival and overall survival were superior for CD23+ MCL compared with CD23-negative MCL
(Kelemen K et al: Am J Clin Pathol 130:166–177, 2008)
.
Other indolent lymphomas recognized by the WHO classification, but not by the Working Formulation, include lymphoplasmacytic lymphoma, splenic marginal zone B-cell lymphoma, extranodal marginal zone lymphoma of MALT type, and nodal marginal zone B-cell lymphoma.
The WHO classification has recognized marginal zone lymphomas (MZLs) as unique clinical and pathologic entities, ie, extranodal (MALT), nodal, and splenic NHL subtypes. MALT NHL is extremely indolent and presents as localized stages I to II disease, infrequently disseminating. The stomach is the most frequent site, but low-grade NHLs (and former pseudolymphomas) of the lungs, thyroid, salivary gland, and orbit are of this type.
Savage and colleagues recently reported outcomes among 181 patients with ALCL. Patients with anaplastic lymphoma kinase-negative ALCL had inferior overall survival compared with patients with ALK-positive (ALK+) ALCL (49% vs 70%, respectively) but had improved overall survival compared with peripheral T-cell NHL (49% vs 20%, respectively). Further, the IPI predicted differential outcomes for both ALK+ and ALK-negative ALCL
(Savage KJ et al: Blood 111:5496–5504, 2008)
.
Staging and prognosis
Determining the extent of disease in patients with NHL provides prognostic information and is useful in treatment planning. However, histologic subclassification (WHO classification) is the primary determinant of survival and potential for cure. Compared with patients with limited disease, those with extensive disease usually require different therapy, and certain extranodal sites of involvement, such as the CNS and testes, require specific treatment modalities.


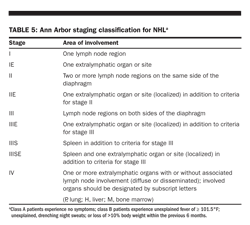
Ann Arbor system Although initially devised for Hodgkin lymphoma, the Ann Arbor system has been routinely applied to NHL (Table 5). Because Hodgkin lymphoma commonly spreads via contiguous lymph node groups, this system is based primarily on the distribution of lymphatic involvement with respect to the diaphragm and the presence of extralymphatic organ involvement. The Ann Arbor system does not reflect the noncontiguous nature of disease spread in NHL, does not discriminate well between stages III and IV disease, and fails to account for tumor bulk or number of extranodal sites.
Some trials in Burkitt and Burkitt-like lymphoma use the St. Jude/Murphy staging system, in part to more completely describe the extent of extranodal disease. Unlike the current WHO classification, this staging system recognizes Burkitt leukemia as a separate entity. Moreover, this system was developed when surgery was used for diagnostic and therapeutic purposes. Patients are also typically stratified into two risk groups, with low-risk patients defined as having a normal LDH level and a single focus of disease measuring less than 10 cm and all others considered to be high risk.
Prognostic factors Histology and morphology remain the major determinants of treatment outcome and prognosis, but gene expression signatures are likely to be the principal determinants in the future. Some patients with slow-growing low-grade lymphoma may remain well for many years with minimal or no initial therapy, whereas survival of patients with some types of high-grade lymphoma is measured only in weeks unless aggressive treatment is initiated promptly. The biologic and clinical behaviors of these disorders vary among the different histologic subtypes.
The International Prognostic Index (IPI) was developed by 16 institutions and cooperative groups



in the United States, Europe, and Canada as a prognostic factor model for aggressive NHL treated with doxorubicin-containing regimens. Clinical features that have been independently predictive of survival are included in Table 6 below.
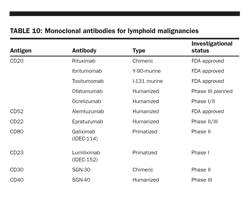
This index appears to be a useful guide for selecting treatment for patients with aggressive, diffuse large cell NHL, by identifying subsets of patients in whom intensified primary therapy may be warranted. Because younger and older patients have markedly different prognoses and younger patients are more likely to be considered for more intensive investigational regimens, an age-adjusted model for patients ≤ 60 years old has been proposed. In younger patients, stage (III or IV), high LDH level, and nonambulatory performance status are independently associated with decreased survival. In the post-rituximab (Rituxan) era, persons with no risk factors have a predicted 5-year overall survival of 94%, compared with 55% for high-risk patients with three to five risk factors.
The IPI also appears to be useful in predicting outcome in relapsed or refractory large B-cell lymphoma patients undergoing autologous stem-cell transplantation (SCT).



A prognostic factor model has been devised based on a study of 919 cases of follicular lymphoma, known as the Follicular Lymphoma IPI (FLIPI; Table 7). Multivariate analysis showed that age (> 60 vs ≤ 60 years), Ann Arbor stage (III− IV vs I− II), number of extranodal sites (> 4 vs ≤ 4), LDH level (above normal vs normal/below normal), and hemoglobin level (< 120 g/L vs ≥ 120 g/L) were predictors of overall survival.
An analysis of the FLIPI in the post-rituximab era has been reported. Multivariate analysis showed that elevated beta2-microglobulin, longest nodal diameter over 6 cm, bone marrow involvement, anemia, and age over 60 years independently predicted survival. Including all patients (n = 832) with 0, 1− 2, or 3− 5 factors, the 3-year PFS was 91%, 69%, and 51% (P = .00001), while the 5-year OS was 79%, 51%, and 20%, respectively (P = .00001). Among patients treated with rituximab-containing regimens only (n = 559), the FLIPI-2 remained predictive of outcome (3-year PFS of 89%, 73%, and 57%, respectively; P = .001).
Immunobiologic factors Various immunobiologic factors have been suggested as predictors of outcome in NHL.
Immunophenotype Several studies have suggested that patients with aggressive T-cell NHL have a higher relapse rate and decreased overall survival than do patients with B-cell disease. These observations have been confirmed in updated REAL/WHO studies involving large numbers of patients.
Tumor cell proliferation Studies using the Ki-67 antibody, a marker of nuclear proliferation, have shown that increased tumor cell proliferation is a poor prognostic factor in diffuse large cell lymphoma and mantle cell lymphoma.
Cytogenetic abnormalities and oncogene expression Mutations of p53 are associated with histologic transformation in follicular NHL, which is a phenomenon frequently associated with a poor prognosis. Expression of bcl-2 in diffuse large cell lymphoma has also been associated with inferior survival, whereas bcl-6 expression is a marker of germinal center derivation, a predictor of a favorable outcome with CHOP (cyclophosphamide, doxorubicin, vincristine [Oncovin], prednisone) like therapy. Dual expression of bcl-2 and c-myc in B-cell lymphomas is also a poor prognostic sign.
The Lymphoma/Leukemia Molecular Profiling Project Group examined the gene expression profile in pretreatment biopsy specimens from 414 patients with DLBCL treated with CHOP or R-CHOP. They identified two gene expression signatures that predicted survival, termed stromal-1 and stromal-2. Similar to prior data in follicular lymphoma, the genes defining the prognostic signatures were expressed not in the tumor cells but in the nonmalignant tumor-infiltrating cells
(Lenz G et al: N Engl J Med 359:2313–2323, 2008)
.
Molecular profiling DNA microarray technology for gene expression profiling has identified distinct prognostic subgroups in DLBCL and follicular NHL. Studies in DLBCL have characterized patients into the following subgroups: germinal center B-like DLBCL, activated B-like DLBCL, and a heterogeneous subgroup termed type-3 DLBCL. In the pre-rituximab era, patients with germinal center B-like DLBCL had a significantly improved overall survival compared with the other molecular profiles. Although in the post-rituximab era, this prognostic difference is much less apparent. Recent studies in follicular NHL have identified two gene expression signatures that also predicted survival: immune-response 1 and immune-response 2. Interestingly, the genes that defined the prognostic signatures were not expressed in the tumor cells but were expressed by the nonmalignant tumor-infiltrating cells (primarily T cells, macrophages, and dendritic cells). A variety of immunologically active cell types including specific T-cell subsets and tumor-associated macrophages, have been associated with prognosis in some studies.
Treatment
The therapeutic approach for NHL differs for each subtype. Chemotherapy remains the most



important modality (Tables 8 and 9). However, in select instances, radiation therapy or surgical resection plays a critical role. Biologic approaches, including interferons, monoclonal antibodies (Table 10), and recombinant toxins, have shown significant activity and are now incorporated into treatment paradigms. Autologous and allogeneic SCTs, traditionally reserved for recurrent or refractory disease, are being evaluated as part of initial therapy in high-risk settings. This section will be organized by NHL subtype to best illustrate the biologic characteristics and therapeutic considerations that determine the management strategy for individual patients. Common NHLs will be covered in depth, whereas less frequent entities will be described in limited detail.
FOLLICULAR LYMPHOMA
Follicular lymphoma comprises 22% of all NHLs; only DLBCL is more common. The clinical presentation may be nodal or extranodal, and bone marrow involvement occurs in the majority of cases. Extensive intra-abdominal adenopathy without peripheral node enlargement is not uncommon. Clinical behavior is variable, reflecting the heterogeneity of the underlying biology; some patients survive decades, whereas others progress rapidly to resistant disease or transform to a more aggressive histology. There are rare spontaneous remissions. Transformation is common, occurring in 3% to 6% of patients each year and ultimately 30% to 50% of all patients. Although generally responsive to treatment, the clinical course of follicular lymphoma is characterized by repeated relapses. Although there was no improvement in survival for patients with follicular lymphoma for many years, there is now evidence that outcomes are improving. Median survival has reached 10 or more years. It is likely that this is, in part, attributable to use of rituximab in combination with chemotherapy, but survival was alr


eady improving before the approval of rituximab.
It is crucial to distinguish between reactive follicular hyperplasia and follicular lymphoma, as the former is a benign condition. Morphologic features as well as the absence of Bcl-2 staining within the follicle and the absence of CD10 and/or Bcl-6 protein expression in the interfollicular areas help to distinguish reactive follicular hyperplasia from follicular lymphoma. Follicular lymphoma is graded according to the number of admixed centroblasts within the neoplastic follicles. Grade 3 follicular lymphoma, previously known as follicular large cell lymphoma, is now subdivided into two subtypes: Grade 3a is characterized by a mixture of centrocytes and centroblasts within the follicle, whereas grade 3b has only sheets of centroblasts with no residual centrocytes. The neoplastic lymphocytes in follicular lymphoma express the pan-B markers CD19, CD20, CD22, and CD79a and antigens of the germinal center (including CD10 and Bcl-6). Most follicular lymphomas express Bcl-2 protein, which is highly correlated with the t(14;18)(q32;q21), the cytogenetic hallmark of the disease. This tra


nslocation results in the juxtaposition of the bcl-2 oncogene into the immunoglobulin H heavy-chain locus on chromosome 14, resulting in its constitutive expression. Follicular lymphoma grade 3b with bcl-6 rearrangement but no t(14;18)(q32;21) may be more closely related to DLBCL than to other follicular lymphomas.
As previously mentioned, the FLIPI is a prognostic index designed specifically for follicular lymphomas based on five adverse prognostic factors (Table 7). Age is the most important factor. Three risk categories have been defined, each consisting of approximately one-third of patients. More than two-thirds of low-risk patients but only one-third of high-risk patients survive 10 years. An alternative system, developed by GELF, focuses on assessing tumor burden by scoring the number of nodal sites, the presence of a mass of 7 cm or larger, the presence of B symptoms, splenomegaly, effusions or ascites, cytopenias, or leukemia. Whereas clinical parameters are surrogates for biologic characteristics, biologic correlates such as gene expression signatures may soon supersede clinical prognostic indicators.
Investigators from the National Cancer Institute (NCI) have described two gene expression signatures associated with vastly different clinical outcomes in follicular lymphoma. These two signatures represent the expression of immunoregulatory genes in the nonmalignant cells infiltrating the malignant lymphoma at the time of diagnosis. Others have reported that the number of tumor-associated macrophages, tumor-infiltrating mast cells, or specific T-cell subsets was an independent predictor of overall survival in follicular lypmphoma, also underscoring the importance of host response in follicular lymphoma.
Treatment of early-stage disease
For the relatively small number of patients with stage I or II follicular lymphoma, radiotherapy continues to be the recommended approach because of the potential for long-term disease-free survival and possible cure. Results from the Princess Margaret Hospital-s series of involved-field radiotherapy (IFRT) for early-stage disease show cumulative relapse rates of 54% and 56% at 15 and 25 years, with only a 2% risk of relapse beyond 15 years. Combined-modality therapy has also resulted in excellent disease control, and a randomized trial comparing IFRT with combined-modality therapy is ongoing.
Despite the excellent outcomes associated with radiotherapy, the National Lymphocare Study revealed that the majority of patients in the United States are either observed or treated with rituximab alone or in combination with chemotherapy, foregoing the potential for cure, even in young patients. Use of functional imaging with PET may improve the results of IFRT by more accurately identifying patients with truly localized disease.
For clinical stages I and IIA low-grade follicular lymphoma, irradiation alone is directed to the entire involved lymphoid region, as defined by Kaplan and coworkers, or the involved region plus one additional uninvolved region on each side of the involved nodes. The recommended dose is approximately 30 Gy for nonbulky disease showing prompt regression and 36 Gy for bulky or slowly regressive disease, in 1.75 to 2.0 Gy daily fractions. As the majority of subsequent relapses occur outside previous radiation fields, often in adjacent or distal lymph nodes, extended-field or total lymphoid irradiation has been used to try to improve cure rates. Clinical series have shown improvement in freedom from relapse only, with no significant difference in long-term survival.
Treatment of advanced-stage diseaseWatch and wait The standard management of asymptomatic patients with follicular lymphoma has been a “watch-and-wait” approach. Treatment is delayed until symptoms or cytopenias intervene or there is impending compromise of vital organs. In the pre-rituximab era, multiple phase III randomized trials comparing immediate chemotherapy with observation for asymptomatic patients with advanced-stage follicular lymphoma have shown no difference in outcome. In fact, for patients older than age 70, the chances of not requiring chemotherapy were 40% at 10 years in a recently published trial. The median time to first systemic therapy for patients randomized to the observation arm was 2.6 years. Complete remission rates, however, were higher in the patients treated immediately after diagnosis than in those who were observed and later treated (63% vs 27%). The achievement of a complete remission may prove to be important if the ultimate goal is to administer postremission therapy (eg, vaccine) that is likely to be most effective in the presence of minimal residual disease. A phase III trial comparing the watch-and-wait approach with rituximab therapy for asymptomatic advanced-stage disease is ongoing in Europe. At the same time, an American intergroup trial comparing two different rituximab dosing regimens is enrolling patients with low tumor burden indolent lymphoma who are asymptomatic.
Irradiation Irradiation for clinical stages III and IV, low-grade, extensive-stage NHL is used locally for palliation of symptomatic sites of disease and is extremely effective. Abbreviated fractionated schedules (25 to 30 Gy in 2.5 to 3 Gy daily fractions, respectively) are often used. A low-dose regimen of 4 Gy in 2 fractions has been shown to be effective, with an overall response rate of approximately 80% in the palliation of symptoms, and is well tolerated. Total-body irradiation, usually consisting of 12 Gy in 2 Gy fractions twice a day, is used as part of preparative regimens for bone marrow transplantation.
Rituximab For patients with symptoms or other reasons for treatment, there are many treatment options, including single or multiagent chemotherapy, monoclonal antibodies or radioimmunoconjugates, combinations of chemotherapy and immunotherapy with anti-idiotype vaccines, and new agents such as bortezomib (Velcade) or bendamustine (Treanda). Treatment with rituximab results in overall response rates of nearly 50%, with a median response duration of approximately 1 year, in relapsed or refractory indolent lymphomas. In previously untreated patients, however, the overall response rate was 80%, and the median progression-free survival is 18 months in one trial from France. Of note, 28% of all treated patients and 34% of all complete and partial responders maintained their responses for 5 or more years. Similar results have been reported by the US NCCTG.
To improve on response rates and duration of response, additional doses of rituximab have been administered as “maintenance therapy.” The median event-free survival is prolonged with this approach, especially for previously untreated patients. In previously treated patients, the total duration of benefit from rituximab appears to be the same whether patients receive maintenance rituximab on a scheduled basis or reinduction with rituximab only at the time of disease progression. A confirmatory trial (ECOG 4402) in untreated patients with a low tumor burden has met accrual, and the results are eagerly awaited. Maintenance regimens have varied, and the impact of the frequency of administration on the duration of response is unknown. Questions also remain regarding the impact of maintenance rituximab on the quality of life and the cost of care.
Rituximab maintenance also impacts progression-free survival among patients treated with chemotherapy alone and those receiving rituximab plus chemotherapy. Compared with observation, maintenance rituximab improved progression-free survival in previously untreated patients with follicular lymphoma who responded to cyclophosphamide, vincristine, prednisone (3-year progression-free survival, 64% vs 33%). Among relapsed/refractory patients treated with CHOP or rituximab-CHOP (R-CHOP) maintenance, rituximab also improved progression-free survival. Whether these results will translate into an overall survival benefit remains to be seen.
Interferon-αThe use of interferon (IFN)-α in follicular lymphoma has been extensively investigated both in combination with chemotherapy and as maintenance therapy, with varying results. In most studies, IFN-αwas associated with a prolongation of remission but not overall survival. A notable exception was the GELF86 trial, in which overall survival was prolonged. The SWOG reported the results of a large phase III trial in which patients with indolent lymphomas were randomized to receive IFN-α or observation following induction with an intensive anthracycline-containing regimen and in some cases radiotherapy. Postremission therapy did not prolong progression-free or overall survival. Although these results, along with the toxicities associated with IFN-α, have led many physicians to abandon its use altogether, a large phase III study is currently ongoing in Germany comparing standard vs intensive dose maintenance.
Chemotherapy with and without rituximab Studies comparing single-agent chemotherapy with multiagent therapy in patients with advanced-stage follicular lymphoma have not shown meaningful differences in outcomes. Fludarabine, identified in the 1980s as an active agent in follicular lymphoma, has been incorporated into combination regimens with high response rates (including molecular remissions) but has not been shown to prolong the duration of remission when compared with other multiagent regimens. Secondary myelodysplastic syndromes and acute leukemias have now been associated with the fludarabine, mitoxantrone, dexamethasone (FND) regimen. High response rates and durable remissions have resulted when rituximab was combined with CHOP chemotherapy in a small number of patients with follicular lymphoma, some of whom were treatment-naive. These encouraging results led to four phase III trials comparing combinations of chemotherapy and rituximab with chemotherapy alone in previously untreated patients. Overall response rates and either median time to treatment failure or event-free survival were superior in the chemoimmunotherapy arm in every series. An overall survival benefit has been demonstrated in three of the four trials and in the high-risk subset of the fourth study.
Consolidation therapy with Y-90 ibritumomab (Zevalin) prolonged progression-free survival from 13.5 months to 37 months in patients with follicular lymphoma in first complete response (CR) or partial remission. Only 14% of the 414 patients enrolled on this randomized phase III trial received a rituximab-containing regimen for induction. Now that the addition of rituximab to chemotherapy has become standard of care based on demonstrated improvements in progression-free and overall survival, the benefit of consolidation with radioimmunotherapy will need to be evaluated in patients treated first with chemoimmunotherapy
(Morschhauser F et al: J Clin Oncol 26:5156–5164, 2008)
.
Radioimmunotherapy The anti-CD20 radioimmunoconjugates Y-90 ibritumomab (Zevalin) and I-131 tositumomab (Bexxar) both deliver ionizing radiation to target cells and their neighbors and have proven to be relatively easy to administer, safe, and effective. Response rates are higher and remissions more durable when radioimmunoconjugates are used early in the clinical course. Both agents are likely to have their greatest impact when used in previously untreated patients.
In previously treated patients, Y-90 ibritumomab, a high-energy beta-emitter, yielded an overall response rate of 80% for relapsed or refractory follicular or transformed CD20+ B-cell NHL, with a median duration of response of 14 months. For patients refractory to rituximab, response rates with Y-90 ibritumomab are high (74% overall response rate), but the median duration of response is relatively short (6.4 months; range, 0.5− 25+ months). The dose-limiting feature of this approach is hematologic toxicity. Short-lived myelosuppression occurs 7 to 9 weeks post treatment. Dosing is based on weight (0.4 mCi/kg), with a reduction (0.3 mCi/kg) for those with mild thrombocytopenia (< 100,000/μL).
I-131 tositumomab is both a gamma- and beta-emitter and is individually dosed on the basis of dosimetry to deliver 75 cGy of total-body irradiation. Similar to Y-90 ibritumomab, it is effective in both heavily pretreated relapsed and refractory patients. Heavily pretreated patients with refractory low-grade or transformed NHL had an overall response rate of 65% (20% complete response rate), with a median duration of response of 6.5 months. These rates were notable in view of a response rate of only 28% in the preceding chemotherapy regimen. Like Y-90 ibritumomab, I-131 tositumomab is associated with predictable myelosuppression. Secondary myelodysplasia and leukemia have occurred in patients treated with radioimmunotherapy, but only in patients previously treated with chemotherapy and thereby already at risk. In previously untreated patients, the complete response rate was 75%, with a 5-year progression-free survival of 59%. These data must be interpreted carefully, as this study enrolled a relatively young patient population (median age, 49 years), with low-bulk disease, a group that some physicians would choose to observe rather than treat.
I-131 tositumomab has been used to consolidate responders following induction with CHOP chemotherapy. In a phase II trial of previously untreated patients with follicular lymphoma, the percentage of complete response/unconfirmed complete response increased from 39% following CHOP chemotherapy to 69% following consolidation with I-131 tositumomab. With a median follow-up of 5.1 years, 66% of patients are alive and disease-free. Based on these phase II results and the encouraging outcome of patients treated with R-CHOP, the American intergroup has recently completed a phase III trial comparing CHOP followed by I-131 tositumomab with R-CHOP in treatment-naive patients. The results of this study will be forthcoming in the near future.
In a plenary presentation at ASCO 2009, Schuster et al presented results from a multicenter phase III study that enrolled 234 patients with previously untreated follicular lymphoma (FL). After chemotherapy with PACE (prednisone, doxorubicin, cyclophosphamide, etoposide) 177 patients who achieved a complete remission were randomized 2:1 to receive patient-specific autologous tumor-derived Id vaccine (BiovaxID) with KLH/GM-CSF or KLH/GM-CSF alone. Complete remission was maintained in 117 patients for greater than or equal to 6 months, and of these patients, 76 were randomized to receive the vaccine (Id-KLH/GM-CSF) and 41 to the control arm (KLH/GM-CSF). At a median follow-up of 56.6 months (range 12.6–89.3 months), the median time to relapse was 44.2 months for the treatment group vs 30.6 months for the control (
P
= .045; HR = 1.6). Investigators concluded that ID vaccination after a chemotherapy-induced remission of greater than or equal to 6 months prolongs remission duration in patients with FL
(Schuster SJ et al: J Clin Oncol 27:18s[Suppl, abstract], 2009.
)
Anti-idiotype vaccines Lymphoma-specific idiotypes serve as tumor-specific antigens in follicular lymphoma and constitute the basis for vaccine therapy. In early vaccine trials, immunized patients who generated an anti-idiotype response experienced longer remissions than those who failed to mount a response. In phase I trials, vaccination resulted in tumor shrinkage in some patients, and in a phase II trial, anti-idiotype vaccine eliminated minimal residual disease detectable only by PCR after intensive chemotherapy. Based on these encouraging results, a phase III randomized trial comparing vaccination plus KLH (keyhole limpet hemocyanin, a nonspecific immunostimulant) to KLH alone in previously untreated patients who achieved a complete remission with intensive anthracycline-containing combination chemotherapy was conducted. Patients receiving the vaccine experienced a statistically significant prolongation of cancer-free survival (see sidebar). In contrast, two other placebo-controlled trials of anti-idiotype vaccination have shown no benefit. Compared with placebo, vaccination following induction chemotherapy with cyclophosphamide, vincristine, prednisone, or induction immunotherapy with rituximab did not impact progression-free or overall survival. Differences in study design are likely responsible for the differences in outcomes among the trials. New directions in vaccination include Id-pulsed dendritic cell and membrane proteoliposomal vaccines.
Novel agents New agents targeting specific molecular targets such as the ubiquitin-proteasome pathway, histone deacetylase, the mammalian target of rapamycin (mTOR), and Bcl-2 have shown promise in the treatment of follicular lymphoma. In recently reported phase II trials, bortezomib has shown activity in follicular lymphoma as well as in mantle cell lymphoma. New combinations including bortezomib are now being investigated. Bendamustine is a novel alkylator with activity in both rituximab-refractory and rituximab-sensitive indolent NHL that has recently been approved by the FDA for relapsed disease that is refractory to rituximab.
Novel antibody approaches to follicular lymphoma include new and improved anti-CD20s, antibodies that bind to alternative targets, and chemoimmuno- and radioimmunoconjugates.
Bendamustine (Treanda) plus rituximab (Rituxan) proved equally effective as rituximab-CHOP in previously untreated patients with follicular, indolent, and mantle cell lymphomas, according to the first interim analysis. In a multicenter randomized phase III trial that enrolled 439 patients, there were no differences in response rates or rates of disease progression or deaths between the two arms, albeit the median observation time was only 17 months. Bendamustine-rituximab was less myelo-suppressive than R-CHOP and overall more tolerable
(Rummel MJ et al: Blood 110: abstract 385, 2007)
.
SCT The natural history of follicular lymphoma is characterized by response to therapy but repeated relapses and progressively shorter and shorter remissions, ultimately resulting in death from progressive disease. Autologous and allogeneic SCTs are alternative strategies often associated with durable remissions that may impact overall survival. Unfortunately, immediate and long-term toxicities are significant and must be considered when assessing the appropriate role of transplantation in the overall treatment plan for individual patients. The only phase III trial to address the role of autologous SCT in patients with relapsed follicular lymphoma closed prematurely because of poor accrual. Nonetheless, progression-free and overall survival rates were significantly longer with high-dose chemotherapy (HDCT) and autologous SCT (with purged or unpurged autografts) than with conventional alkylator therapy. Whether any of the new therapeutic strategies will prove to be as effective as autologous SCT in the relapsed setting remains to be seen.
Compared with conventional-dose rituximab (Rituxan)-CHOP, high-dose sequential (HDS) therapy, including rituximab and autologous stem cell transplant, did not improve overall survival for previously untreated patients with follicular lymphoma in a phase III randomized multicenter trial conducted by the Italian cooperative groups. Although the 4-year event-free survival was 28% for R-CHOP and 61% for HDS (
P
< .001), overall survival was the same in both arms. Patients whose disease progressed after R-CHOP had excellent outcomes after HDS as second-line therapy
(Ladetto M et al: Blood 111:4004–4013, 2008)
.
Several groups have investigated the role of autologous SCT as consolidation therapy for patients in first complete or partial remission. Although progression-free survival may be prolonged, an impact on survival has not been demonstrated consistently. An increased incidence of secondary myelodysplasia following autologous SCT in first remission has reduced enthusiasm for this approach. Contemporary trials evaluating the role of autologous SCT for follicular lymphoma in first remission induced with rituximab-containing regimens are needed. The results of a GLSG trial comparing IFN maintenance with myeloablative chemotherapy with autologous SCT after induction with CHOP or R-CHOP are awaited.
An alternative approach to consolidating complete and partial remissions achieved with conventional induction therapy is the use of a sequential HDCT program, which culminates in an autologous SCT. The Italian cooperative groups have recently reported results of a phase III randomized trial comparing R-CHOP for 6 cycles with a sequential HDCT with autologous SCT. Again, there was a significant difference in event-free survival but no difference in overall survival.
Single-institution studies as well as analysis of registry data suggest that a tumor-free graft is an important determinant of outcome in follicular lymphoma. Administration of rituximab during stem-cell mobilization provides an “in vivo” purge, reducing contamination of the autograft with malignant lymphocytes. The long-term benefit of such an approach has not yet been demonstrated.
Among patients undergoing allogeneic stem cell transplants for follicular lymphoma reported to the Center for International Blood and Marrow Transplant Research, more than 80% were conditioned with reduced-intensity allo-transplants (3-year overall survival; 71% vs 62%, respectively;
P
= .15), but an increased risk of late disease progression was observed after reduced-intensity conditioning
(Hari P et al: Biol Blood Marrow Transplant 14:236–245, 2008)
.
Allogeneic SCT has been investigated primarily in young patients with HLA-identical sibling donors and extensive disease and/or marrow involvement. Low relapse rates suggest that this approach is potentially curative but is associated with high treatment-related morbidity and mortality. Reduced-intensity transplantation is based on the assumption that a graft-vs-lymphoma effect is operative and has the potential to cure follicular lymphoma. Whether this approach will reduce toxicity while maintaining the low relapse rates associated with standard myeloablative allotransplants remains to be established. A randomized trial comparing this strategy with autologous SCT in relapsed follicular lymphoma has been initiated in the United States.
Overall treatment strategy
Whereas treatment choices were once limited to single or combination alkylator-based treatment, we now are faced with choosing among a wide variety of strategies. There are many unanswered questions that can only be addressed through well-designed clinical trials. Hence, whenever possible, every patient with follicular lymphoma should be enrolled in prospective clinical studies. In the absence of symptoms or other indications for treatment, patients should be observed. A combination of rituximab and chemotherapy is recommended in the absence of a clinical trial for those who require treatment. Selected patients with comorbidities may be best served with rituximab alone. Radioimmunotherapy is a good option at the time of relapse, with transplantation reserved for selected patients in first or subsequent relapse.
Chronic lymphocytic leukmia/SMALL LYMPHOCYTIC LYMPHOMA
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is a malignancy of small, round, B lymphocytes involving peripheral blood, bone marrow, and lymph nodes. The term “SLL” is reserved for cases in which there are no circulating malignant lymphocytes. SLL generally presents with lymph node and splenic involvement. Involvement of the bone marrow and peripheral blood may develop later in the course of disease. At the time of presentation, patients may be asymptomatic, complain of only fatigue, or have symptoms related to cytopenias (including autoimmune hemolytic anemia, lymphadenopathy, or splenomegaly). The immunophenotype helps to distinguish CLL/SLL from other B-cell leukemias/lymphomas, including mantle cell and leukemic forms of follicular lymphoma. Typically, the malignant lymphocytes stain weakly with surface immunoglobulin, CD20, CD22, and CD79b; they are CD5+, CD23+, and FMC7− . Cytogenetic abnormalities are detected in the majority of cases when fluorescence in situ hybridization (FISH) analysis is used. Trisomy 12, deletions at 13q14, and deletions at 11q22-23 are common. Many molecular markers of prognosis have been studied in CLL, including Zap-70, but their value in SLL is unknown.
Given the relatively small numbers of patients with SLL, they have generally been included in clinical trials of “indolent lymphoma.” Conventional alkylator-based regimens with rituximab as well as purine analogues and combinations thereof have been used when patients become symptomatic. Anthracyclines have not been shown to benefit patients with CLL/SLL. When compared with follicular lymphoma, CLL/SLL is less likely to respond to rituximab as a single agent. Alemtuzumab (Campath), a potent therapy for CLL, is less effective in treating nodal disease than peripheral blood and bone marrow involvement. SCT, both autologous and allogeneic, has been studied in selected patient populations but should be reserved for relapsed young patients with a good performance status.
Splenic marginal zone lymphoma
Splenic marginal zone lymphoma (SMZL) is a rare disorder comprising less than 1% of NHLs. Clinically, this lymphoma most often presents as splenomegaly with splenic hilar node involvement but without peripheral adenopathy. The bone marrow is commonly involved, and malignant villous lymphocytes may be detected in the peripheral blood. Cytopenias are a common presenting feature, often related to hypersplenism and less frequently to an autoimmune process or marrow replacement. Sometimes confused with CLL or mantle cell lymphoma, SMZL may be distinguished by its immunophenotype. Typically, cells are CD20+, CD79a+, CD5− , CD10− , CD23− , and CD43− . Staining for cyclin D1 is negative, excluding mantle cell lymphoma. The absence of CD103 helps to exclude hairy cell leukemia. Complex karyotypes are common. The clinical course is indolent. Cytopenias respond to splenectomy with long-lasting remissions. High response rates have been reported with rituximab, but the longevity of those responses remains to be determined. Transformation to more aggressive histologies may occur. Fludarabine alone or with rituximab appears to be more effective than alkylators but may be associated with significant toxicity.
Nodal marginal zone lymphoma
Nodal marginal zone lymphoma (NMZL) is a primary nodal B-cell disorder that resembles lymph nodes involved by MZLs of extranodal or splenic origin without extranodal or splenic involvement. Lymphadenopathy (either localized or generalized) is the presenting complaint in most cases. Extranodal lymphoma may be uncovered in the evaluation of many cases of suspected NMZL. The clinical course is usually indolent, similar to that of other MZLs.
Extranodal marginal zone B-cell lymphoma of MALT type
MALT lymphomas comprise only 7% to 8% of B-cell lymphomas but nearly 50% of all gastric lymphomas. Although the GI tract is most often involved, other common sites include the lungs, head and neck, ocular adnexae, skin, thyroid, and breasts. There often is an associated history of autoimmune disorders, such as Sjgren-s syndrome or Hashimoto-s thyroiditis or chronic inflammatory processes secondary to infectious agents (H pylori, B burgdorferi, or C psittaci). A form of MALT involving the small bowel (immunoproliferative small intestinal disease, previously known as α-chain disease) has been associated with C jejuni. The majority of patients present with stage I or II disease. The frequency of bone marrow involvement appears to differ depending on the primary site of involvement. Multiple extranodal sites may be involved at the time of presentation. Transformation to a high-grade lymphoma may occur in approximately 8% of cases.
The malignant lymphocytes of MALT lymphoma are typically CD20+, CD79a+, CD5− , CD10− , and CD23− . The t(11;18)(q21;q21) is characteristic of MALT lymphomas, particularly those involving the stomach or lungs. The translocation creates a fusion between the MALT-1 gene, which is an essential regulator of bcl-10− mediated NF (nuclear factor)-kB signaling, and the API2 gene, which inhibits apoptosis. This genetic abnormality is a marker of MALT lymphomas that do not respond to antibiotic therapy for H pylori infection, are associated with a more advanced stage, and do not transform into more aggressive histologic subtypes. This translocation has not been associated with nodal or splenic MZLs or other types of lymphoma. Additional characteristic translocations have been discovered (Table 1), but their clinical significance is uncertain at this time.
Treatment of H pylori infection with triple therapy (eg, omeprazole [Prilosec], metronidazole, and clarithromycin [Biaxin]) results in regression in the majority of early lesions. However, tumors invading beyond the submucosa and lesions with t(11;18) are associated with a failure to respond to H pylori eradication, deep penetration, and distant spread.
Localized MALT gastric lymphoma that does not respond to antibiotics may be cured with local irradiation, with a field including the stomach and perigastric lymph nodes. This treatment is safe, extremely effective, and preserves the stomach. A single-institution experience reported a 96% complete response rate and a 90% freedom-from-treatment-failure rate, at a median follow-up of 4 years. If local irradiation fails, chemotherapy or rituximab, and in some instances surgery, can be used. Alkylator-based therapy or purine analogs have been used with success for persistent or disseminated disease. The typical dose of radiotherapy is 30 Gy in 20 fractions directed to the stomach and perigastric lymph nodes. Localized nongastric MALT lymphomas also respond well to local radiotherapy.
Lymphoplasmacytic lymphoma/Waldenstrm's macroglobulinemia
Lymphoplasmacytic lymphoma/Waldenstrm's macroglobulinemia is a disorder of small B lymphocytes; plasmacytoid lymphocytes; and plasma cells, typically involving the bone marrow, lymph nodes, and spleen. It is usually associated with a serum monoclonal protein (usually IgM) with associated hyperviscosity or cryoglobulinemia. The clinical presentation is usually related to hyperviscosity with visual symptoms, stroke, or congestive heart failure. Peripheral neuropathies occur in approximately 10% of patients related to reactivity of IgM with myelin-associated glycoprotein or gangliosides. An association with HCV infection has been demonstrated. Characteristically, the immunophenotypic analysis reveals surface and cytoplasmic immunoglobulin, usually IgM type, and B-cell− associated antigens (such as CD19, 20, 22, and 79a). The malignant cells are CD5− , CD10− , and CD23− .
The clinical course is generally indolent. Asymptomatic patients may be observed. Plasmapheresis may be appropriate first therapy for those who present with hyperviscosity. The clinical status of the patient, not the level of the protein, determines when treatment is initiated. Choice of therapy depends on many individual factors, including age, comorbidities, and the particular indication for therapy. Rituximab and nucleoside analogues (cladribine and fludarabine) as well as the traditional oral alkylators have shown efficacy. Combinations of these agents are also under study. Bortezomib, thalidomide (Thalomid), and alemtuzumab have shown activity in Waldenstrm-s macroglobulinemia. SCT, both autologous and allogeneic, is being investigated in younger patients with relapsed or refractory disease.
Diffuse large b-cell lymphoma
Clinical presentation DLBCL makes up about one-third of the cases of NHL and is classified as a mature peripheral B-cell neoplasm by WHO. The clinical presentation is variable, but generally patients present with either peripheral lymphadenopathy (neck, axillae) or enlarged nodes in the mediastinum, the mesenteric region, or the retroperitoneum. These sites predict symptoms, which may include chest pain; facial swelling and suffusion of the eyelids (superior vena cava [SVC] syndrome from mediastinal disease); abdominal discomfort, ascites (mesenteric), or back pain; or renal obstruction (retroperitoneal presentations). More than 30% of patients present with disease in extranodal sites, such as the GI tract (including Waldeyer-s ring), skin, bone marrow, sinuses, GU tract, thyroid, and CNS. B symptoms, consisting of fever, sweats, and weight loss, are more common in DLBCL than in the indolent lymphomas and occur in about 30% of patients. The median age at presentation is 60 years.
Once the diagnosis is clearly established, staging studies are carried out to determine treatment and define parameters for follow-up. Generally, imaging studies of the chest, abdomen, and pelvis are obtained, and CT scans provide the most accurate anatomic information. Recently, functional imaging using PET scans (which have largely replaced gallium scans) has shown promise as a means of distinguishing between residual scar and active disease after treatment. Further, some investigators have shown that early response by PET scan (after 2 to 3 cycles) is a good prognostic indicator. In addition to CT and PET scans, bone marrow aspirate and biopsy, serum LDH level, and serum beta-2 microglobulin level have been described as important predictors of outcome.
Pathology/immunology The diagnosis should be made by incisional or excisional biopsy of an available lymph node, with adequate tissue for immunologic studies, such as flow cytometry or immunohistochemistry (IHC), to identify the characteristic B-cell clonality (kappa or lambda restriction). In many cases of DLBCL, CD10 is present, indicating a germinal center origin. The CD20 antigen is present in almost all cases of B-cell lymphomas and on almost all normal B cells. In addition, markers for bcl-2 and bcl-6 offer prognostic information and are part of most diagnostic evaluations of DLBCL. The use of FNA or core biopsy should be discouraged and is acceptable only when tissue cannot be safely obtained by other means and only if flow cytometry is used to help classify the disease and distinguish it from epithelial malignancies that can masquerade as lymphoma.
Prognostic factors Clinical predictors of response have been identified and are now widely used to


help design therapeutic plans and clinical trials. These predictors include patient age (< or > age 60), performance status (0, 1 vs 2− 4), number of extranodal sites (more than two), Ann Arbor stage (I or II vs III or IV), and serum LDH level (> normal; Table 6). Older patients, higher stage, poorer performance status, higher number of extranodal sites, and higher LDH level all predict a worse outcome, and this model has been validated in more than 3,000 patients. These parameters have been called the IPI; this index is used to plan therapy and clinical trials in the United States and abroad and may be used to predict survival (Table 11).
More recently, genomics have been used to help predict outcome based on molecular signature (see subsection on Molecular profiling). This molecular system provides prognostic information independent of the IPI. Several investigators using similar statistical methodologies have yielded comparable results, and recently these analyses have been extended to other lymphomas.
Treatment Prior to the 1970s, most patients with stage I/II large cell lymphoma (intermediate grade in the Working Formulation) were treated with irradiation alone, with overall cure rates of 40% to 50%. Patients with pathologically favorable stage I/II disease had even better outcomes, but relapse rates, even in these patients, were still 20% to 30%. Pathologic staging, therefore, selected a group suitable for irradiation alone. This approach is no longer appropriate, in view of the success of combined chemotherapy and irradiation in clinically staged patients.
Coiffier et al found that the addition of rituximab improved results in elderly patients with DLBCL, and recent data confirm these observations for younger patients as well. For patients with clinical stage I or II disease (by the Ann Arbor criteria), most studies suggest that chemotherapy (CHOP and most would add rituximab) for 3 to 4 cycles followed by localized radiation therapy is preferred. Excellent local and systemic control is obtained with combined-modality therapy.
In an ECOG phase III trial, Horning et al showed that 8 cycles of CHOP and irradiation produced a 10-year disease-free survival rate of 57%, compared with 46% with CHOP alone (P = .04). Overall survival was 64% vs 60%, respectively (P = .23), and time to disease progression was 73% vs 63%, respectively (P = .07).
Miller et al showed that CHOP (3 cycles of CHOP and irradiation) produced a progression-free survival at 5 years of 77%, vs 64% for 8 cycles of CHOP alone (P = .03). Overall survival at 5 years was 82% vs 72%, respectively (P = .02). A recent update of this SWOG study was reported by Miller et al, with an 8.2-year median follow-up. The 5-year estimates for CHOP (3 cycles plus irradiation) vs CHOP (for 8 cycles) remained unchanged. Kaplan-Meier estimates now show overlapping curves at 7 years for failure-free survival and 9 years for overall survival. The treatment advantage for CHOP (for 3 cycles plus irradiation) for the first 7 to 9 years was diminished because of excess late relapses and NHL deaths occurring between 5 and 10 years. Patients with good IPI risk factors had a 5-year overall survival of 94%; patients with one adverse risk factor had an overall survival of 70%; those with three adverse risk factors had a 5-year survival of 50%.
These results were confirmed by a single-arm (doxorubicin-containing chemotherapy) approach followed by IFRT conducted by the British Columbia Cancer Agency. However, two reports from European investigators question the value of consolidation irradiation in early-stage disease. These studies did not use rituximab or 18F-fluorodeoxyglucose (FDG)-PET staging, and details on the irradiation technique used were not available. The necessity of consolidation radiation therapy after complete response to R-CHOP chemotherapy is now being tested in a randomized study in Germany.
Until further studies define the optimal therapy for stages IA to IIA DLBCL (nonbulky), many investigators consider 3 to 4 cycles of R-CHOP and IFRT the initial treatment of choice. For patients with bulky disease, a minimum of 6 cycles of R-CHOP is typically administered. Irradiation doses of 30 to 36 Gy, delivered in 1.75 to 1.8 Gy over 3 to 4 weeks after completion of systemic therapy, appear to be adequate. Radiation fields usually include involved lymph node sites or an involved extranodal site and its immediate lymph node drainage areas. Furthermore, the disease should be easily encompassed in a radiation field with acceptable toxicity.
Disease site or potential toxicities may influence the treatment plan:
• Lymphomas of the head and neck may be managed with chemotherapy alone to avoid the acute mucositis and long-term xerostomia associated with radiation therapy fields that are large and include both parotid glands. Alternatively, precise radiation therapy techniques can be employed with intentional sparing of salivary glands, using intensity-modulated radiation therapy (IMRT).
• Fully resected gastric or small intestinal lymphoma may be treated with chemotherapy alone. Patients at high risk of perforation or life-threatening hemorrhage may require surgical resection. Alternatively, chemotherapy followed by local irradiation allows gastric preservation and is preferred in most patients.
For patients with more advanced stage (III or IV) disease, CHOP has been the standard (now with rituximab) for 6 to 8 cycles or 2 cycles beyond remission. Recent data suggest an advantage to “dose-dense” therapy, shortening the interval between cycles from 3 to 2 weeks with growth factor support. More data are needed to validate these results. Many studies now suggest an advantage to the addition of immunotherapy in the form of rituximab, and in almost every study, the combination of rituximab and chemotherapy has improved the response rate and disease-free survival. There appears to be no advantage to maintenance therapy with rituximab in this setting, however, as long as rituximab is included in the induction. Responses are seen in upward of 80% of patients, and approximately 50% to 60% achieve a complete remission. It appears that 50% of these patients (30% to 50% overall) are likely cured.
For patients who either do not have a complete remission or who relapse, alternative therapies are possible, but long-term responses have been seen mostly with autologous or allogeneic SCT. Patients who do not have responsive disease prior to SCT generally do poorly. The IPI has been used to predict outcome for transplantation in DLBCL. The role of autologous SCT for high-risk patients remains open to debate. A randomized clinical trial of early vs delayed high-dose therapy for patients with high- and high-intermediate risk diffuse aggressive lymphoma conducted by the US intergroup is just completing accrual. If this trial confirms the benefit of early SCT in poor-risk patients with chemosensitive diffuse aggressive NHL, subsequent studies will focus on increasing the number of patients who become eligible for transplant consolidation. Investigational treatments include novel antibodies, radioimmunotherapy, and single-agent chemotherapy drugs. Nonmyeloablative SCT is being evaluated in patients with recurrent or refractory disease.
Some investigators believe that irradiation for stages III and IV (advanced or extensive) DLBCL may be added after the completion of definitive chemotherapy if there is localized residual disease to improve local tumor control. Irradiation may also be delivered after chemotherapy to areas of initially bulky disease, again to enhance local tumor control. These recommendations are based on the observation that when DLBCL relapses after definitive chemotherapy, it usually does so in initially involved or bulky areas of disease. The benefits and potential side effects of irradiation should be weighed against the use of alternative chemotherapy salvage regimens.
Mantle cell lymphoma
Clinical presentation By comparison with low-grade NHL, patients with mantle cell lymphoma are older (median age is 64 years), mostly male (75%), and more likely to have peripheral blood involvement (about 30%) and extranodal involvement (mostly the GI tract and CNS, as discussed below). The clinical course in mantle cell lymphoma is characterized by the worst features of the aggressive lymphomas (an aggressive course) and the indolent lymphomas (frequent recurrences). The disease is often widespread at diagnosis, and marrow involvement and splenomegaly are common. GI tract involvement is also common, and many centers suggest evaluation of the GI tract at the time of diagnosis. CNS recurrences are frequent (up to 20%), but isolated CNS disease is a rare occurrence. Leukemic presentations have been described.
Pathology/immunology This disorder was originally classified as diffuse, small, cleaved lymphoma by the REAL classification and represents less than 10% of all NHLs. In this disease, a homogeneous population of small lymphoid cells with irregular nuclear borders arises from and expands the mantle zone surrounding the germinal centers of the lymph nodes, spreading diffusely through the node as the germinal centers are overrun. The lymphocytes express IgM or IgD, as in CLL, but in much greater density. It was recognized that the cells carry a translocation of the long arms of chromosomes 11 and 14 notated as t(11;14)(q13;q23). This molecular event juxtaposes the bcl-1 gene on chromosome 11 to the immunoglobulin heavy-chain gene on chromosome 14, leading to overexpression of bcl-1. This gene encodes the cell-cycle regulatory protein cyclin D1, which is believed to play a role in checkpoint control in DNA synthesis. The immunophenotype is characteristic, and mantle cell lymphomas are usually CD5+, CD20+, CD10− , CD23− , and FMC7+. This immunophenotype is similar to that seen with CLL/SLL, except that CD23 is most often expressed in CLL/SLL but usually is not expressed in mantle cell lymphoma. The key to the diagnosis is the demonstration in tumor tissue or peripheral blood of the t(11;14) by FISH or the cyclin D1 protein by IHC. Inactivation of the ATM gene has been described in mantle cell lymphoma. Recent data show that mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d), suggesting a relationship with the stromal environment.
Treatment Responses to aggressive chemotherapy (CHOP or R-CHOP) are seen, but patients relapse frequently, and median survival is short. Recent data suggest an advantage to SCT while patients are in remission, but more data are required to validate these results. Patients who have relapsed after autologous SCT have been “rescued” by allogeneic SCT.
The National Comprehensive Cancer Network (NCCN) guidelines recommend clinical trials for



patients with mantle cell lymphoma (there is no “standard” therapy), and ongoing trials are investigating a short course of R-CHOP followed by radioimmunotherapy with ibritumomab. The complete response/unconfirmed complete response rate is 55% after R-CHOP plus ibritumomab, compared with 13% after R-CHOP alone; however, further follow-up is needed. Investigators have found that aggressive hyperfractionated chemotherapy with rituximab (R-hyper-CVAD) may result in long-term responses in patients with mantle cell lymphoma (Table 12). Anti-idiotype vaccine studies are also under way in this disease, and novel chemotherapy regimens/agents designed to take advantage of the molecular biology of this disease (flavopiridol, bortezomib, cladribine) are being tested. In addition, recent data suggest that a combination of rituximab and the novel hybrid alkylating agent bendamustine produces high response rates in patients with recurrent mantle cell lymphoma.
Recently, the Nordic group presented data that suggest it is possible to achieve long-term disease-free survival in patients with mantle cell lymphoma. They compared their prior experience (1996− 2000) using a high-dose CHOP regimen for 4 cycles followed by an autologous SCT with BEAM (carmustine [BiCNU], etoposide, cytarabine [Ara-C], methotrexate) or BEAC (carmustine [BiCNU], etoposide, cytarabine [Ara-C], cyclophosphamide) conditioning with a newer regimen (2000− 2006). With the newer regimen, patients received Ara-C in between cycles of CHOP, and then rituximab was added starting at week 10. This regimen was followed by an autologous SCT with BEAM or BEAC. Patients were then followed using molecular markers by PCR (t[11;14]); those who showed molecular recurrence were treated with rituximab preemptively. Only morphologic relapse was considered a “relapse” for the purposes of this trial.
The results of the two trials were compared and showed that 5-year event-free survival improved from 15% to 41% and overall survival, from 63% to 75%. These data suggest that there may be a plateau in the curve in mantle cell lymphoma and that the addition of Ara-C and rituximab may have contributed to that improvement. Further studies are needed to verify these data. Recently, Martin et al from Cornell have suggested that patients with mantle cell lymphoma can be observed without treatment. Better performance status and lower-risk IPI scores were characteristic of the patients who could be observed. This finding suggests that a deferred approach to treatment is acceptable in a select group of patients with mantle cell lymphoma.
In addition, new agents such as lenalidomide and nutlin-3 have been found to have activity in mantle cell lymphoma.
An update and summary of the German Mantle Cell Lymphoma group have recently been published, and these data suggest that overall, the outlook for patients with this disease has improved.
Burkitt and Burkitt-like lymphoma
Clinical presentation These diseases present as three distinct clinical entities: endemic, sporadic, and immunodeficiency-related types. Endemic Burkitt lymphoma most often presents in young children or adolescents with large nodes in the neck, often involving the maxilla or mandible. These cases are most often seen in equatorial Africa and follow the distribution of endemic malaria, hence its designation as “endemic Burkitt lymphoma.” In American, or sporadic, Burkitt or Burkitt-like lymphomas, the disease presents in the abdomen and extranodal sites, especially in the GI tract. Sporadic Burkitt lymphoma accounts for 1% to 2% of all adult lymphomas in Western Europe and the United States. The immunodeficiency type is seen in the setting of HIV infection but can be seen in patients with CD4 cell counts > 200 cells/μL. In both endemic and sporadic Burkitt and Burkitt-like lymphomas, males are affected more often than females.
The LDH level is often elevated, owing to the high turnover rate of these cells and the bulk of disease. The bone marrow and CNS are often involved, and if not involved initially, they are at risk, so CNS prophylaxis is needed. A staging system for Burkitt and Burkitt-like lymphomas has been developed by Murphy and associates.
Pathology/immunology These lymphomas are the most rapidly proliferating NHLs. Under the microscope, it is difficult to distinguish Burkitt from Burkitt-like lymphomas and from the B-cell French-American-British (FAB) L3 variant of acute lymphoblastic leukemia. Indeed, the WHO classification recognizes the lymphoma and leukemic phases as a single entity, a mature B-cell neoplasm. The disease is characterized by medium-sized cells with an abundant basophilic cytoplasm with lipid vacuoles. There are round nuclei with clumped chromatin and multiple nucleoli; a diffuse pattern of infiltration is seen and is classic for Burkitt lymphoma. The numerous macrophages that are usually seen in the lymph node biopsy specimens give rise to the so-called starry-sky appearance.
The proliferative rate of this tumor is high, and there are frequent apoptotic cells. In the Burkitt-like variant, there is greater pleomorphism in nuclear size and shape, and the nuclei have fewer nucleoli. There is a low level of concordance among pathologists (about 53%) when they attempt to distinguish Burkitt from Burkitt-like lymphomas, and even by clinical criteria, that distinction is difficult. The cells express surface IgM, CD19, CD20, CD22, CD10, and CD79a and do not express CD5, CD23, and TdT. Bcl-6, a zinc finger protein, is usually expressed. The major consideration in the differential diagnosis is precursor B-cell lymphoma/leukemia, which in contrast expresses TdT; surface immunoglobulin is mostly negative. CD20 may also be negative in this disorder. In Burkitt lymphoma, the expression of CD10 and Bcl-6 protein suggests that these cells originate from the germinal center, and indeed, this is confirmed by sequence analysis of the immunoglobulin variable heavy-chain and light-chain genes. Somatic hypermutation of these genes has been described.
Genetics The almost constant genetic abnormality in Burkitt lymphoma is overexpression of the c-myc oncogene; in 80% of cases, this abnormality results from a balanced translocation between chromosomes 8 and 14, notated as t(8;14), where the c-myc oncogene on chromosome 8 is juxtaposed to immunoglobulin heavy-chain enhancer elements on chromosome 14. In the remaining 20% of cases, there are other translocations, including t(2;8)(p12;q24) and t(8;22)(q24;q11). There have been different breakpoints identified in Burkitt lymphoma, and they have been associated with the sporadic and immunodeficiency subtypes.
EBV One cannot discuss these highly aggressive NHLs without discussing the role of EBV. This virus, a member of the herpesvirus family, has the ability to infect resting B cells and transform them into proliferating blasts, most likely by bypassing antigens on lymphocytes and activating signaling molecules. By contrast, certain viruses (HCV) and bacteria (H pylori) may cause lymphoma by activating lymphocytes in an antigen-specific manner. EBV infection results in a polyclonal proliferation of lymphoblasts that are latently infected with the virus, as opposed to the infection seen in infectious mononucleosis, which is a lytic infection. This process is regulated by the expression of up to nine latent viral proteins, which are under the control of the transcription factor EBV nuclear antigen 2 (EBNA-2). It appears that the type and result of EBV infection in lymphoid tissue are controlled by various “growth programs,” each causing expression of different viral proteins, which then determine the fate of the infected cell. These in vitro events are different from what occurs in healthy carriers of EBV (up to 90% of the population have been exposed), where the viral proteins are not expressed because all of the latently infected cells are resting memory B cells. It is in the germinal center of the lymph node, however, that virally infected cells can transform into memory B cells, as the viral proteins are expressed within the B cells of the germinal center.
Although EBV was found in patients with Burkitt lymphoma over 40 years ago, the role of EBV in the disease still remains uncertain. The exact role of c-myc overexpression in the pathogenesis of the disease is also not known, but c-myc is known to play a role in cell-cycle progression and cellular transformation. EBV is found in over 95% of cases of “endemic Burkitt lymphoma,” which occurs in Africa, but its role in the pathogenesis of the disease is still not clear. The reason therapy with antiviral agents (ganciclovir or acyclovir) cannot be used to treat EBV-associated lymphomas is that the required thymidine kinase (TK) gene is not expressed in latent EBV infection. Recent studies using the small molecule arginine butyrate to upregulate the TK gene and protein expression with concomitant antiviral antibiotics have met with some success.
Treatment Patients must be treated quickly after diagnosis, which should be made on a full biopsy so that adequate tissue is obtained. Tumor lysis syndrome occurs most often with Burkitt lymphoma, and attempts to reduce uric acid production with allopurinol or to degrade it with the enzyme rasburicase (Elitek) should be part of the management, as should aggressive hydration. Patients should be managed in a facility with access to support such as urgent dialysis, because it may be necessary if tumor lysis syndrome occurs.
Treatment includes aggressive chemotherapy, with anthracyclines and cyclophosphamide as the


cornerstone. Regimens incorporating hyperfractionated cyclophosphamide such as hyper-CVAD, developed by Murphy and adopted by the M. D. Anderson group, have been used. Other published regimens include CODOX-M/IVAC (cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, and high-dose Ara-C; Table 13) and the French regimen, which incorporate intensive therapy given weekly in various combinations and intrathecal chemotherapy, and systemic high-dose methotrexate or high-dose Ara-C to facilitate CNS penetration. In children, the results are excellent, and about 80% of patients can be cured. In adults, the outcome is not as favorable, but with newer more intensive regimens, 40% to 60% of patients survive 5 years without disease. New approaches using rituximab and early SCT are being investigated, as are new agents (flavopiridol and an analog of resveratrol) with unique mechanisms of action, which may have relevance to Burkitt and Burkitt-like lymphoma. Addition of rituximab to hyper-CVAD has resulted in marked improvement in outcome in adults with Burkitt lymphoma.
Primary mediastinal large B-cell lymphoma
Clinical presentation Primary mediastinal large B-cell lymphoma (PMLBCL) occurs most often in young women (female:male ratio is 2:1) who present with mediastinal masses only. These masses are usually bulky and often invade surrounding structures, such as the pleura, lungs, pericardium, and chest wall, but disease is infrequently found outside the chest cavity. At recurrence, however, extranodal sites such as the lungs, adrenal glands, liver, or kidneys may be involved. Because of the location and bulk of the disease, patients complain of chest pain, cough, and shortness of breath and are often found to have SVC syndrome. This can be subtle, with unexplained breast enlargement the only symptom in some cases. The diagnosis can be delayed if the clinician does not recognize the signs and symptoms of SVC. This clinical presentation is similar to classic Hodgkin lymphoma, and indeed, that is the primary differential diagnostic consideration when these patients are evaluated.
Pathology/immunology The pathology is characterized by a diffuse proliferation of large cells with clear cytoplasm, often accompanied by extensive sclerosis. The cells are mostly of B-cell origin and express CD20 and other B-cell markers but do not express surface immunoglobulin (Ig). Indeed, the discordant expression of CD79a and Ig expression are distinguishing features of PMLBCL. There are data that describe gains of chromosomal material in tissue specimens, most often 2p, 9p, 12q, and Xq. The rel, mal, and fig1 (interleukin-4 [IL-4] gene) oncogenes are overexpressed in tissue specimens. Ig genes have a high level of somatic hypermutation. All of these observations suggest that this entity is unique, especially compared with B-cell lymphomas that arise in peripheral nodes. IL-13 expression and downstream effectors of IL-13 signaling pathways are overexpressed, along with tumor necrosis factor (TNF) family members and TNF receptor-associated factor-1.
The overexpression of the rel oncogene, previously described, has been associated almost exclusively with the nucleus, consistent with NF-κB pathway activation, and the mal overexpression has been confirmed in gene array studies. These data help us to reorder our thinking about these clinically unique lymphomas and to begin to build a molecular story that is consistent with the clinical observation that PMLBCL is more like classic Hodgkin lymphoma than like DLBCL. Further, the observations that certain signaling pathways are involved provide a rationale to attack these pathways specifically in a targeted approach.
Treatment The clinical course is variable; some report a poor outcome with conventional, CHOP-based chemotherapy regimens and irradiation, and some report an excellent outcome. It seems clear that bulk of disease and LDH level are important prognostic factors and that prediction of the outcome by the IPI is useful. A variety of chemotherapy regimens have been evaluated, including CHOP and MACOP-B/VACOP-B (methotrexate or etoposide, Adriamycin, cyclophosphamide, Oncovin, prednisone, bleomycin), and more recently rituximab has been incorporated into the management. Usually, radiation therapy is a part of the initial treatment; however, recent data suggest that radiation therapy may not be necessary in all patients. In general, in 2009, patients receive anthracycline-containing chemotherapy with rituximab, and after 4 to 6 courses, radiation therapy may be given to patients with bulky disease. There are no randomized trials comparing radiation therapy with no radiation therapy in this setting. PET scanning may influence the use of consolidation radiation therapy in the future.
Peripheral T-cell lymphoma, unspecified
Peripheral T-cell lymphoma, unspecified (PTCL) is predominantly a nodal lymphoma that represents the most common T-cell lymphoma subtype in Western countries, comprising approximately 50% to 60% of T-cell lymphomas and 5% to 7% of all NHLs. PTCL usually affects male adults (1.9:1 male-to-female ratio) with a median age of 61 years (range, 17− 90), with 25% of patients presenting in stage I or IIE; 12%, in stage III; and 63%, in stage IV. Patients with PTCL from this study commonly presented with unfavorable characteristics, including B symptoms (40%), elevated LDH level (66%), bulky tumor ≥ 10 cm (11%), nonambulatory performance status (29%), and extranodal disease (56%), leading to the majority of patients (53%) falling into the unfavorable IPI category (score of 3 to 5).
Most T-cell NHL patients are treated in the same manner as aggressive B-cell patients, with anthracycline-based combination chemotherapy such as CHOP. Randomized trials comparing CHOP with other combination regimens confirmed CHOP as a standard regimen for aggressive B-cell NHL; unfortunately, these trials do not allow for subset analysis of T-cell patients. Rituximab should not be included in the treatment of PTCL (unless other conditions such as immune thrombocytopenic purpura exist), as CD20 is not expressed. Other therapeutic agents being tested in T-cell NHL include purine and pyrimidine analogs, denileukin diftitox (Ontak), and a retinoic acid/IFN-αcombination.
Aviles and colleagues completed a randomized trial of CHOP vs CMED (
see Table 8
) for 217 untreated peripheral T-cell lymphoma NOS patients. In an intent-to treat analysis, complete remission rate for CMED was 76% vs 57% for CHOP. Moreover, with 10.5-year median follow-up, progression-free survival and overall survival rates for CMED were 63% and 75%, respectively, compared with 26% and 38%, respectively for CHOP (
P
< .01;
Aviles A et al: Med Oncol 25:360–364, 2008)
.
Denileukin diftitox is a novel recombinant fusion protein consisting of peptide sequences for the enzymatically active and membrane translocation domains of diphtheria toxin with recombinant IL-2 (CD25 receptor); it has been studied mostly in cutaneous T-cell NHL, although clinical benefit has been reported in other T-cell NHL patients. Recently, the histone deacetylase inhibitors suberoylanilide hydroxamic acid (SAHA) and depsipeptide have shown activity against PTCL. In addition, the Office of Oncology Drug Products recently granted accelerated approval to pralatrexate injection (Folotyn, Allos Therapeutics, Inc.) for the treatment of patients with relapsed or refractory PTCL (see sidebar below).
Angioimmunoblastic T-cell lymphoma
Angioimmunoblastic T-cell lymphoma (AITL), also known as angioimmunoblastic lymphadenopathy with dysproteinemia, is one of the more common T-cell lymphomas, accounting for 15% to 20% of cases and 3% to 4% of all lymphomas. Pathologically, AITL has distinct features, with a diffuse polymorphous infiltrate, prominent arborizing blood vessels, perivascular proliferation of follicular dendritic cells, and the presence of large B-cell blasts often infected with EBV. The malignant cells are mature follicular helper CD4 αΒ T cells. The mean age at presentation is 57 to 65 years, with a slight male predominance, and the majority of patients present with stage III or IV disease. AITL is commonly a systemic disease with nodal involvement with various associated disease features, such as organomegaly, B symptoms (50%− 70%), skin rash, pruritus, pleural effusions, arthritis, eosinophilia, and varied immunologic abnormalities (positive Coombs- test, cold agglutinins, hemolytic anemia, antinuclear antibodies, rheumatoid factors, cryoglobulins, and polyclonal hypergammaglobulinemia).
O-Connor and colleagues reported results using the novel antifolate, pralatrexate, for the treatment of relapsed/refractory lymphoma. The overall response rate (ORR) was 31% including a 17% CR rate. The ORR was higher in T-cell compared with B-cell lymphomas (54% vs 10%, respectively) and all CRs were seen in T-cell lymphoma patients
(O-Connor OA et al: J Clin Oncol 27:4357–4364, 2009)
.
Spontaneous disease regression is seen on rare occasions, although AITL typically follows an aggressive clinical course. Treatment with anthracycline-based combination chemotherapy results in complete remission rates of 50% to 70% of AITL patients, although only 10% to 30% of patients are long-term survivors. One prospective, nonrandomized multicenter study treated newly diagnosed “stable” AITL patients with single-agent prednisone and combination chemotherapy for relapsing/refractory patients or initially if “life-threatening” disease was present at diagnosis. The complete remission rate was 29% with single-agent prednisone, whereas the complete remission rate for relapsed/refractory patients or patients treated initially with combination chemotherapy was 56% and 64%, respectively. With a median follow-up of 28 months (range, 7 to 53), the overall and disease-free survival rates were 40.5%: 24%− 56%) and 32.3% (CI: 17%− 47%), respectively, although the median overall survival was 15 months.
There are anecdotal reports of relapsed AITL patients who have responded to immunosuppressive therapy, such as low-dose methotrexate/prednisone, as well as reported responses to purine analog treatment. Furthermore, cyclosporine has demonstrated activity in relapsed AITL patients in case reports, and the ECOG is evaluating this agent in a prospective study. There are anecdotal reports of thalidomide (Thalomid) plus steroid responses in AITL.
Anaplastic large-cell lymphoma, T-/null-cell, primary systemic type
Anaplastic large cell lymphoma (ALCL), primary systemic type, is a CD30-positive T-cell lymphoma that accounts for approximately 2% to 3% of all NHLs. This disease mainly involves lymph nodes, although extranodal sites may be involved (not exclusively the skin; see subsection on ALCL, CD30+ cutaneous type). This disease may be divided in part based on the expression of the tyrosine kinase anaplastic lymphoma kinase (ALK), created from a balanced chromosomal translocation t(2;5) and other less common translocations involving 2p23 (see Table 1). When heterogeneous patient populations are analyzed, the prevalence of ALK positivity in primary systemic ALCL cases is 50% to 60%. ALK-positive ALCL is typically diagnosed in men prior to age 35 (male-to-female ratio, 1.7:1), with frequent systemic symptoms and extranodal and advanced-stage disease. ALK-negative patients are usually older (median age, 61 years), with a male-to-female ratio of 1.5:1, with a similar high incidence of extranodal disease.
In addition to the prognostic importance of ALK positivity, the IPI has been identified as an independent prognostic factor within the group of ALK-positive ALCL patients, with a reported 5-year overall survival of 94% vs 41% for IPI 0 or 1 and 2 to 4, respectively. This better prognosis is apparent despite ALK-positive patients more commonly presenting with a poorer performance status and more advanced-stage disease compared with ALK-negative patients.
Therapy for pediatric ALCL is often based on prognostic risk factors, with treatment regimens modeled after high-grade B-cell NHL protocols. Following a brief cytoreductive prephase, short, intensified polyagent chemotherapy is administered, with the number of cycles dependent on the stage of disease. Therapy for adult ALCL, systemic type, has commonly included anthracycline-based regimens such as CHOP. Autologous hematopoietic SCT in first complete remission for ALK-negative ALCL has been advocated by some groups, although this approach warrants prospective validation.
Hepatosplenic T-cell lymphoma
Hepatosplenic T-cell lymphoma (HSTCL) is an uncommon T-cell lymphoma that is seen mainly in young males (median age, 35) presenting with B symptoms, prominent hepatosplenomegaly, mild anemia, neutropenia, thrombocytopenia (commonly severe), significant peripheral blood lymphocytosis, and rare lymphadenopathy and is associated with an aggressive clinical course (median survival, 12 to 14 months).
The tumor cells are usually negative for CD4 and CD8 (85%); positive for CD2, CD3, and CD7 (negative for CD5); and express CD56 in 70% to 80% of cases. TIA-1 is present in almost all cases, but commonly granzyme B and perforin are not present, an indication of a nonactivated cytotoxic T-cell phenotype. Cells usually express the γ/δ T-cell receptor (Vd1+/Vd2− /Vd3−) but are negative for EBV.
Historically, patients with HSTCL have been treated with CHOP-like regimens. Early autologous SCT has been favored by some investigators based on anecdotal cases; however, if feasible, an allogeneic transplant may be more appropriate. A recent report described activity with the purine analog pentostatin (Nipent) in relapsed HSTCL patients. Approximately 10% to 20% of HSTCL cases arise in immunocompromised patients, predominantly in the solid-organ transplant setting.
Extranodal NK/T-cell lymphoma and nasal-type
Extranodal NK/T-cell lymphoma, and nasal-type, formerly known as angiocentric lymphoma, is rare in Western countries, being more prevalent in Asia and Peru. The disease commonly presents in men at the median age of 50 years. This entity is associated with EBV and is typically characterized by extranodal presentation and localized stage I/II disease but with angiodestructive proliferation and an aggressive clinical course. These tumors have a predilection for the nasal cavity and paranasal sinuses (“nasal”), although the “nasal-type” designation encompasses other extranodal sites of NK/T-cell lymphomatous disease (skin, GI, testis, kidneys, upper respiratory tract, and rarely orbit/eye).
Combined-modality therapy incorporating doxorubicin-based chemotherapy (minimum of 6 cycles for patients with stage III or IV disease), IFRT (minimum 50 Gy), and intrathecal prophylaxis are recommended for patients with extranodal NK/T-cell lymphoma, nasal, although the benefit of the addition of chemotherapy to radiation therapy has not been confirmed for limited-stage disease.
Patients with systemic disease have poor long-term survival (5-year overall survival, 20% to 25%), with high locoregional (over 50%) and systemic failure rates (over 70%). L-asparaginase (Elspar) has recently been shown to have significant activity against this lymphoma.
Enteropathy-type intestinal T-cell lymphoma
Enteropathy-type intestinal T-cell lymphoma (EITCL; also known as intestinal T-cell lymphoma) is a rare T-cell lymphoma of intraepithelial lymphocytes that commonly presents with multiple circumferential jejunal ulcers in adults with a brief history of gluten-sensitive enteropathy. EITCL accounts for less than 1% of NHLs, according to the ILSG, and has been recognized to have a poor prognosis, with reported 5-year overall and disease-free survival rates of 20% and 3%, respectively. This finding is in part related to many patients presenting with a poor performance status and varied complications of locally advanced disease by the time a diagnosis of EITCL has been confirmed.
EITCL may present without an antecedent celiac history, but most patients have abdominal pain and weight loss. Evidence of celiac serologic markers such as positive antigliadin antibodies and/or HLA types such as DQA1*0501/DQB1*0201/DRB1*0304 may be present at diagnosis of EITCL. Moreover, these genotypes may represent celiac patients at higher risk for development of EITCL. Small bowel perforation or obstruction, GI bleeding, and enterocolic fistulae are recognized complications of this disease. The immunophenotype consists of pan− T-cell antigens, usually CD8+, and the mucosal lymphoid antigen CD103 is often expressed.
Following diagnosis of EITCL, doxorubicin-based combination chemotherapy should be considered for each patient, and aggressive nutritional support with parenteral or enteral feeding is critical in the care of these patients. Patients with known celiac disease should adhere to a gluten-free diet.
Adult t-cell leukemia/lymphoma
The retrovirus HTLV-1 has been documented to be critical to the development of ATLL. HTLV-1 is known to cause diseases other than ATLL, including tropical spastic paraparesis/HTLV-1− associated myelopathy, infective dermatitis, and uveitis. In endemic areas in Japan, approximately 10% to 35% of the population is infected with HTLV-1. Among these carriers, the overall risk of ATLL is approximately 2.5% in patients who live to age 70. Of the Caribbean population, 2% to 6% are HTLV-1 carriers, whereas less than 1% of the population in lower-risk areas, such as the United States and Europe, are seropositive. HTLV-1 is transmitted through sexual intercourse, transfused blood products (products containing white blood cells, not fresh frozen plasma), shared needles, breast milk, and vertical transmission. Transfusion of HTLV-1− contaminated blood products results in seroconversion in approximately 30% to 50% of patients, at a median of 51 days.
The clinical features of 187 ATLL patients included a median age at onset of 55 years, lymphadenopathy (72%), skin lesions (53%), hepatomegaly (47%), splenomegaly (25%), and hypercalcemia (28%) present at diagnosis. The differential diagnosis between cutaneous ATLL and mycosis fungoides is often difficult. ATLL is separated into four subtypes divided by clinicopathologic features and prognosis: acute, lymphoma, chronic, and smoldering. Shimoyama and colleagues reported on the characteristics of 818 ATLL patients. Patients with acute-type ATLL present with hypercalcemia, leukemic manifestations, and tumor lesions and have the worst prognosis, with a median survival of approximately 6 months. Patients with lymphoma-type ATLL present with low circulating abnormal lymphocytes (< 1%) and nodal, liver, splenic, CNS, bone, and GI disease; the median survival is 10 months. Patients with the chronic type present with > 5% abnormal circulating lymphocytes and have a median survival of 24 months, whereas the median survival of patients with the smoldering type has not yet been reached.
ATLL is an aggressive neoplasm with resistance to conventional chemotherapy, in part due to the viral protein tax-mediated resistance to apoptosis and overexpression of p-glycoprotein (the product of the multidrug resistance-1 gene). Patients may initially respond to combination chemotherapy, but unfortunately, response durations are brief (5 to 7 months). El-Sabban and colleagues combined arsenic trioxide (Trisenox) with IFN-α, which induced cell-cycle arrest and apoptosis. Response rates of 70% to 90% to combination IFN-α and zidovudine therapy have been demonstrated in ATLL, with associated increased median survival rates compared with those of historic controls (11 to 18 vs 4 to 8 months, respectively). The Japanese Clinical Oncology Group Study randomized untreated aggressive ATLL to VCAP-AMP-VECP chemotherapy vs biweekly CHOP. They reported a higher CR rate and improved survival rates with VCAP-AMP-VECP although treatment-related toxicity was high.
Other agents with anecdotal activity in ATLL include irinotecan and the purine analogs (pentostatin and 2-chlorodeoxyadenosine [Cladribine, Leustatin]), although pentostatin did not appear to improve outcomes when added to combination chemotherapy. Future research should include the investigation of recombinant toxins and antibodies, such as denileukin diftitox and alemtuzumab. Allogeneic SCTs have also been incorporated into the treatment strategies.
Cutaneous T-cell lymphomas
Cutaneous T-cell lymphomas (CTCLs) constitute a group of cutaneous NHLs with clonal expansion


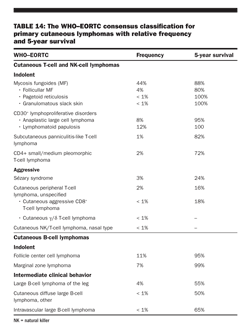
of T lymphocytes into the skin. Several entities are recognized by the combined EORTC and WHO classification, which is based on morphologic, histopathologic, and molecular features (Table 14). The frequency and disease-specific survival differ for each entity.
Mycosis fungoides/Szary syndrome
Mycosis fungoides and its variants represent the most common type of CTCL, comprising 50% of CTCLs, with a male predominance of approximately 2:1 and a predominance of African-American patients of 1.6:1. It has a yearly incidence of 0.36 cases per 100,000 population that has remained constant over the past decade. Clinical and histologic diagnosis of mycosis fungoides has proved to be difficult, because in early stages, it may resemble other dermatoses such as eczematous dermatitis, psoriasis, and parapsoriasis.
Clinically, mycosis fungoides is characterized by erythematous patches, evolving into plaques or tumors; however, the progress is variable. It is classified as an indolent lymphoma by the EORTC.


The neoplastic cells have a mature CD3+, CD4+, CD45RO+, CD8− memory t-cell phenotype. Szary syndrome is the aggressive, leukemic, and erythrodermic form of CTCL, which is characterized by circulating, atypical, malignant T lymphocytes with cerebriform nuclei (Szary cells), and lymphadenopathy. Circulating Szary cells also have a mature memory T-cell phenotype with loss of CD7 and CD26. For staging purposes, the tumor node metastasis (TNM) system is most commonly used (Table 15).
Investigative and approved options that have shown activity against mycosis fungoides/Szary syndrome include the histone deacetylase inhibitors vorinostat (Zolinza, FDA approved), depsipeptide, and panobinostat; the proteasome inhibitor bortezomib; and monoclonal antibodies targeting CD4 (zanolimumab), CD2 (siplizumab), and CD30 (SGN-30).
Treatment of early-stage disease
At present, CTCLs are regarded as incurable. In early CTCL, the cell-mediated immune response is usually normal. Therefore, the majority of these cases can be treated successfully with topical modalities. Early aggressive therapy does not improve the prognosis of patients with CTCL. The skin-targeted modalities include psoralen plus ultraviolet A (PUVA); narrow-band− ultraviolet B (NB-UVB); skin electron-beam radiation therapy; spot radiation therapy; as well as topical preparations of steroids, retinoids, carmustine, or nitrogen mustard (Table 16). Radiation therapy prescriptions may be similar to those for other lymphomas or may be delivered at high dose in limited fractions.
Treatment of advanced-stage disease
A limited number of patients progress to more aggressive and advanced disease with either cutaneous or extracutaneous tumor manifestations. Treatment goals in advanced stages should be to reduce the tumor burden, relieve symptoms, and decrease the risk of transformation into aggressive lymphoma. Established treatment options include mono- or polychemotherapy including COP (cyclophosphamide


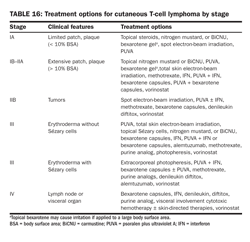
, vincristine [Oncovin], prednisone) or CHOP regimens, extracorporeal photopheresis, interferons, retinoids and rexinoids (bexarotene [Targretin] capsules), histone deacetylase inhibitors (vorinostat and romidepsin [Istodax]), monoclonal antibodies (alemtuzumab), and recombinant toxins (denileukin diftitox). Combinations are frequently used (Table 16). Select patients with progressive and recalcitrant disease have been cured with an allogeneic SCT.
Primary cutaneous cd30-positive Lymphoproliferative disorders
Primary cutaneous CD30-positive lymphoproliferative disorders are the second most common group of CTCL, representing approximately 30% of CTCLs. This spectrum of diseases includes lymphomatoid papulosis, ALCL CD30+ cutaneous type, and borderline cases. The distinction between these entities can be challenging and is often made on clinical behavior.
Lymphomatoid papulosis
Lymphomatoid papulosis is most commonly associated with mycosis fungoides, CD30+ large T-cell lymphoma, and Hodgkin lymphoma. Three histologic types have been identified, characterized as types A, B, and C. Types A and C consist of large lymphocytes resembling Reed-Sternberg cells. Type A cells are embedded in a dense inflammatory background, whereas type C cells form large sheets imitating CD30+ large T-cell lymphoma. Type B simulates classic features of mycosis fungoides, with epidermotropism and a dermal band-like infiltrate composed of small to medium cells. Lymphomatoid papulosis lesions occasionally exhibit clonal gene rearrangements.
Lymphomatoid papulosis represents a benign, chronic recurrent, self-healing, papulonodular, and papulonecrotic CD30+ skin eruption. However, 10% to 20% of patients may develop a lymphoid malignancy, but the prognosis for patients with lymphomatoid papulosis is otherwise excellent, with a 5-year survival of 100%. There is no curative treatment available. Lymphomatoid papulosis is managed by observation, intralesional steroid injection, topical bexarotene, ultraviolet light therapy, or low-dose methotrexate.
Anaplastic large cell lymphoma, CD30+ cutaneous type
Primary systemic CD30+ ALCL and primary cutaneous CD30+ ALCL represent identical morphologic entities, but they are clinically distinct diseases.
The neoplastic cells of primary cutaneous CD30+ ALCL are of the CD4+ helper T-cell phenotype with CD30 expression. It represents 9% of CTCLs and typically presents with solitary or localized nodules. This tumor has an excellent prognosis, as confirmed in several studies, in contrast to the transformation of mycosis lymphoma to a CD30− large cell variant. It shows histologic and immunophenotypic overlap with lymphomatoid papulosis. In most cases, tumor cells show anaplastic features, less commonly a pleomorphic or immunoblastic appearance. However, there is no difference in the prognosis and survival rate. Primary CD30+ ALCL rarely carries the t(2;5) translocation and is usually ALK-negative. These lesions may undergo spontaneous regression, as do the lesions of lymphomatoid papulosis. The mechanism of tumor regression remains unknown.
Spot radiation therapy or surgical excision is the preferred treatment, with systemic chemotherapy reserved for cases with large tumor burden and extracutaneous involvement. More recently, there has been reported efficacy of recombinant IFN-γ -1b (Actimmune) and combined treatment with bexarotene and IFN-α-2a (Roferon-A).
SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA
Subcutaneous panniculitis-like T-cell lymphoma (SCPTCL) is a rare T-cell lymphoma that infiltrates the subcutaneous fat without dermal and epidermal involvement, causing erythematous to violaceous nodules and/or plaques. Systemic symptoms are frequent and include weight loss, fever, and fatigue. The disease may be complicated by the hemophagocytic syndrome. SCPTCL may be preceded by a benign-appearing panniculitis for years. The infiltrate is pleomorphic and associated with inflammation and necrosis. The T-cell phenotype is α/+ with CD4(+/− ), CD8+, and CD56(− /+). Standard treatment has historically included CHOP-like chemotherapy. However, recent data suggest patients can be controlled for long periods with local radiation treatment and/or steroids. Five-year survival rates exceed 80%.
CUTANEOUS γ/δ T-CELL LYMPHOMA
Cutaneous γ/δ T-cell lymphoma is a rare panniculitis presenting with disseminated (ulcerated) plaques, nodules, or tumors. Involvement of mucosal or extranodal sites is common. Systemic symptoms, including weight loss, fever, and fatigue, are almost always present. The hemophagocytic syndrome is often noted. The γ/δ+ T cells are characteristically CD2+, CD3+, CD4− , CD5− , CD7(− /+), CD8(− /+), and CD56+. Aggressive chemotherapy is indicated, with consideration of autologous or allogeneic SCT incorporated into the initial treatment schema. The median survival is less than 2 years.
Pleomorphic T-cell lymphomas with small/medium cells
The small/medium pleomorphic CTCL type appears clinically with single erythematous to violaceous nodules or tumors and accounts for less than 3% of CTCL cases. Most cases have an unfavorable prognosis, with a median survival of ≤ 24 months; however, the CD3+, CD4+, CD8− , CD30− subtype with limited lesions might be associated with a better prognosis, with a reported 45% 5-year survival rate.
The optimal therapy for pleomorphic T-cell lymphomas with small/medium cells has not been defined. Localized lesions have been treated with radiation therapy or surgical excision. Only short-term outcome has been reported. Patients with generalized skin disease or progression have been treated effectively with systemic treatments, including multiagent chemotherapy, retinoids, interferons, and monoclonal antibodies.
Cutaneous B-cell lymphomas
Primary cutaneous B-cell lymphomas (CBCLs) are rare entities. They constitute up to 25% of all cutaneous lymphomas. However, the incidence of CBCLs has been underestimated due to the absence of immunologic and molecular markers. In addition, their terminology and classification remain controversial, with until recently separate and distinct terminology promoted by WHO and EORTC. Primary CBCLs are distinct from nodal lymphomas, and the majority of them have an excellent prognosis. Several types are recognized, with the most common types being follicle center cell lymphoma and marginal zone lymphoma.
EORTC and ISCL consensus recommendations for the management of CBCLs were recently published
(Senff NJ et al: Blood 112:1600–1609, 2008)
.
Follicle center cell lymphoma Follicle center cell lymphoma (FCCL) is defined as a proliferation of centrocytes (small to large cleaved cells) and centroblasts (large round cells with prominent nuclei), showing a nodular or diffuse infiltrate in the majority of cases and presenting only rarely a true follicular pattern. FCCL is the most common subtype, comprising 40% of CBCLs. FCCL shows a predilection for the head, neck, and trunk in elderly patients, with a median age of 60 years and a male predominance of approximately 1.5:1. The clinical course is usually indolent, with an excellent overall survival of up to 97%. However, relapses occur frequently. The large round cell morphology might be associated with a higher rate of disease progression and poorer prognosis.
Small centrocytes predominate in low-grade FCCL, whereas an increased number of large cells occurs in high-grade FCCL; however, lesions with pure high-grade disease may behave indolently and should not by themselves drive the treatment administered. In contrast to their nodal counterpart, bcl-2 is usually not expressed in neoplastic cells, and the t(14;18) translocation is rarely detected. More recently, low rates of bcl-2 expression have been reported. In addition to CD10+ and bcl-6+ expression, FCCL also has an aberrant expression of CD45 RA and CD43 and thus provides a helpful clue to distinguish it from pseudolymphomas. Radiation therapy is often the preferred therapy for solitary or localized group lesions. Surgical excision can be considered for small lesions. Chemotherapy, though effective, rarely results in cure. Rituximab has proven to be effective for palliation. Observation is a reasonable alternative in many instances.
Immunocytoma /marginal zone lymphoma Immunocytoma (IC)/marginal zone lymphoma (MZL) is a recently recognized low-grade lymphoma and represents the second most common subtype of CBCLs. It predominantly occurs on the upper and lower extremities. The median age at presentation is 55 years, and females are affected more often than males. The reported survival rates are 97% to 100%, although relapses commonly occur. Histologically, IC/MZL has features of MALT lymphomas and shows a nodular or diffuse dermal infiltrate with a heterogeneous cellular infiltrate of small lymphocytes, lymphoplasmacytoid cells, plasma cells, intranuclear inclusions (Dutcher bodies), and reactive germinal centers that may be infiltrated by neoplastic cells. Diagnosis can be difficult, because of the variable composition of the infiltrate that may be interpreted as a reactive process or as FCCL. In contrast to FCCL, MZL is negative for bcl-6 and CD10. In 50% of cases, CD43 is highly expressed. Large cell transformation and a head and neck presentation may be associated with a worse prognosis. Therapeutic alternatives are similar to those described for FCCL.
Large B-cell lymphoma of the leg
Primary cutaneous large B-cell lymphoma of the leg (LBCLL) forms a separate category in the WHO− EORTC classification, as a more aggressive type seen in elderly patients, with a median age of 76 years at diagnosis and a female predominance of 7:2. Most cases have a follicle center cell origin, and histologic evaluation shows a diffuse dermal infiltrate with predominance in large B cells with multilobulated nuclei, comprised of centroblasts and immunoblasts, with presence of small, cleaved cells and a minor admixed infiltrate component. Eosinophilic intranuclear (Dutcher body) or intracytoplasmic (Russell body) inclusions of immunoglobulin are common. Unlike FCCL, LBCLLs consistently express bcl-2, although they are not associated with the t(14;18) translocation.
The prognosis is less favorable for LBCLL than for other CBCLs, with a 5-year survival rate of 50% to 60%. Prognostic factors identified with a poor outcome include the predominance of round cells (centroblasts/immunoblasts) over cleaved cells (centrocytes) in the tumor infiltrate, MUM-1 expression, and multiple lesions at presentation. The use of an IPI-based model is required to investigate whether LBCLL is associated with a poorer prognosis. These lymphomas should be treated as systemic DLBCLs with anthracycline-based chemotherapy. In patients presenting with a single, small skin tumor, radiotherapy is a consideration. Rituximab has also been incorporated into combination regimens.
Intravascular large B-cell lymphoma WHO and EORTC have proposed intravascular large B-cell lymphoma (IVLBCL) or angiotropic B-cell lymphoma as a provisional entity. This subtype is rare and corresponds to the proliferation of malignant lymphocytes within lumina of small vessels, involving most frequently the skin and CNS. It was previously considered a vascular tumor and referred to as malignant angioendotheliomatosis. Although the majority of cases are of B-cell origin, few cases of T-cell lineage have been reported. The reason for intravascular localization is not clear, but association with an unknown surface receptor or dysfunction of lymphocyte-endothelial interaction affecting adhesion molecules has been suspected.
IVLBCL is clinically characterized by tender erythematous, purpuric, indurated patches and plaques located on the trunk and thighs, where it can resemble panniculitis. Cases of generalized telangiectasia over normal skin have been reported. Cytomorphology reveals intravascular occlusion of small vessels, filled with large atypical centroblast-like B lymphocytes. IHC shows CD19, CD20, CD45, and CD79a expression. Genotypic analysis has demonstrated clonality, although it may not be positive in every case. Generally, the prognosis of this aggressive type of lymphoma is poor despite the use of combination chemotherapy, because of the initial or secondary CNS involvement. Prognosis appeared better in some reports, if isolated cutaneous involvement was present. However, no large series permitting a precise prognosis to be determined are available.
cd4+/CD56+ Hematodermic Neoplasm (blastic nk-cell lymphoma)
The CD4+/CD56+ hematodermic neoplasm commonly presents in the skin, with nodular and extracutaneous systemic involvement. This rare disorder appears to be derived from a plasmacytoid dendritic cell precursor. T-cell receptor genes are in germline configuration. This entity causes a dismal prognosis (median survival, 14 months) despite intensive chemotherapy.
Plasmacytoma
Primary cutaneous involvement of plasmacytoma is uncommon and generally develops as a consequence of direct spread from an underlying multiple myeloma. It represents 4% of extramedullary plasmacytomas and affects predominantly elderly men, with a median age of 60 years at diagnosis. It is characterized by a monoclonal proliferation of mature plasma cells. Cutaneous plasmacytomas are potentially curable, with a 5-year survival rate of > 90%. However, the presentation of multiple lesions is an important adverse prognostic factor. Histopathology shows a dense monomorphous dermal infiltrate of plasma cells with a varying degree of maturation and atypia, admixed with few lymphocytes and histiocytes. Neoplastic plasma cells express clonal immunoglobulin, CD38, and CD79a but are negative for CD20. Rarely, amyloid deposition within the tumor is demonstrated, which is more common in secondary cutaneous involvement of plasmacytoma. A recent organized workshop on plasma cell dyscrasias questioned whether these cases are true cutaneous plasmacytomas, represent reactive B-cell infiltrates associated with an infectious etiology, or represent a variant of MZL with a predominant population of plasma cells. Diagnosis may rely on demonstration of monoclonality by restriction of Ig light-chain expression. Excision or radiation treatment is most commonly used.
HIV-RELATED LYMPHOMAS
Most lymphomas seen in patients who have HIV infection are of an aggressive histology and advanced stage at presentation. Extranodal disease is common, with unusual sites of presentation, including the GI tract, CNS, and multiple soft-tissue masses. Some patients present with primary CNS lymphoma. Poor-risk factors include a high LDH level, large tumor bulk, extranodal disease, and low CD4 cell counts (< 100 cells/μL). Because of their increased risk of opportunistic infections and impaired hematologic reserve, historically many patients with HIV-related lymphomas have been unable to tolerate aggressive chemotherapy regimens. Current antiviral medications have allowed for the use of more traditional regimens, including R-CHOP and R-EPOCH (rituximab, etoposide, prednisone, vincristine [Oncovin], cyclophosphamide, doxorubicin [hydroxydaunorubicin]), with results comparable to those of other NHL patients with similar histologies and presentations.
CNS prophylaxis with intrathecal chemotherapy is necessary to prevent meningeal dissemination. (For a more detailed discussion of HIV-related NHL, see chapter 24.)
Posttransplantation NHL
Posttransplantation lymphoproliferative disorders (PTLDs) remain one of the most morbid complications associated with solid organ transplantation (SOT). The incidence varies from 1− 2% in renal transplant recipients to 12− 14% in heart transplant recipients, the latter who require more potent immunosuppressive therapy.
The pathologic spectrum of PTLD is heterogenous, comprising a spectrum ranging from hyperplastic appearing lesions to frank aggressive lymphoma. The majority of cases are classified as monomorphic subtype, of which the most common histology is akin to DLBCL. PTLDs are fully depicted in the updated WHO classification (see Table 4).
Historically, PTLDs were reported to occur at a median of 6 months from SOT, although recent data suggests this interval may be longer (ie, median 40− 60 months). Early PTLD cases (ie, < 12 months after SOT) more often express EBV, whereas late-onset cases are typically EBV-negative.
Overall survival rates have been poor with mortality rates ranging from 50%− 70% in most studies. However, recent evidence suggests improved outcomes in the modern era (see sidebar).
Treatment for PTLD initially involves the reduction of immunosuppression (usually by at least 50%), especially for early EBV-positive cases. EBV-negative PTLD will respond to immunosuppression reduction but less frequently (15%− 25%) than EBV-positive cases (50%− 65%). In addition, the status of the transplanted organ will in part dictate the amount of immunosuppression safely allowable to avoid organ rejection. The exact role of rituximab in B-cell PTLD is not well defined. Single-agent rituximab was evaluated in two phase II studies for patients who failed RI with reported overall response rates of 42% and 73% with modest survival rates. However, a recent retrospective series using frontline rituximab-based therapy, in conjunction with reduced immune suppression, was associated with significantly improved survival compared with prior reports (see sidebar).
A Chicago multicenter collaboration investigated a cohort of 80 PTLD patients consecutively treated over a recent 10-year period. The majority of patients (80%) received rituximab-based treatment, most (74%) as a component of frontline therapy together with reduced immune suppression. With a median SOT-to-PTLD time of 42 months (1-216), the 3-year PFS for all patients was 57% with 3-year OS of 62%. Patients who received rituximab-based therapy as part of initial treatment had 3-year PFS of 70% and OS 73% compared with 21% (
P
< .0001) and 33% (
P
= .0001), respectively, without rituximab. On multivariate regression analysis, three factors were associated with progression and survival: CNS involvement, bone marrow involvement, and hypoalbuminemia. A survival model based on these factors was formed
(Evens AM et al: J Clin Oncol 27:15s [Suppl, abstract], 2009)
.
The decision of initially treating with rituximab alone vs rituximab with chemotherapy (eg, R-CHOP) is often determined on a patient-by-patient basis. Factors in small studies associated with lower response to rituximab in PTLD include EBV-negative disease and elevated LDH. In addition, chemotherapy may be needed as initial therapy for patients who have a large tumor burden warranting rapid response of disease. Of note, during chemotherapy, immunosuppressant medication doses should be either significantly reduced or carefully stopped completely, to avoid infectious complications. Carefully selected patients with relapsed/refractory monomorphic PTLD are able to receive high-dose chemotherapy followed by autologous SCT, with long-term survival reported in some series.
Anecdotal reports have described the activity of TK inhibitor antiviral therapies such as ganciclovir and acyclovir to prevent and/or treat PTLD, although the data are not convincing. This finding is not surprising, as EBV survives as an episome outside the lymphocyte genome, and these drugs do not eradicate latently infected B cells. However, one group has shown that arginine butyrate was able to induce EBV tyrosine kinase activity in EBV-immortalized B cells and convert patient-derived latently infected B-cell lymphoma tumor cells that were resistant to ganciclovir to a sensitive phenotype. A phase I/II study with encouraging clinical results was recently reported.
Primary CNS lymphoma
Primary CNS lymphoma is a rare form of NHL, arising within and confined to the CNS. Histologically, primary CNS lymphomas are indistinguishable from systemic NHLs. More than 40% of patients have evidence of leptomeningeal dissemination, and 15% have ocular disease at presentation. A stereotactic needle biopsy is the procedure of choice for diagnosis. Resection does not appear to improve survival.
The two most important prognostic factors in primary CNS lymphoma are age (> 50 years) and poor performance status (Karnofsky performance score < 70). Retrospective studies have documented that treatment of primary CNS lymphoma with whole-brain radiotherapy (WBRT) alone (with or without corticosteroids) results in a median survival of 10 to 15 months, with a 5-year survival of 3% to 4%. Current standard therapy for newly diagnosed primary CNS lymphoma is systemic chemotherapy, including high-dose intravenous methotrexate-based therapy (at least 2,500 mg/m2 per cycle) alone or more commonly combined with agents that penetrate the CNS (vincristine, procarbazine [Matulane], and high-dose cytarabine). This regimen results in median survival rates of 50 to 60 months. The most important component in the treatment of primary CNS lymphoma is the use of high-dose methotrexate therapy (at least 2,000 mg/m2). WBRT had been considered a standard component following chemotherapy; however, long-term neurotoxicity remains a major concern, especially for patients older than 60 years.
A recent study reported encouraging results for primary CNS lymphoma patients who achieved complete remission following 5 to 7 cycles of high-dose methotrexate-based chemoimmunotherapy and subsequently received reduced-dose WBRT (23.4 Gy) compared with the standard WBRT of 45 Gy. Long-term survival data are available using autologous or allogeneic SCT for relapsed/refractory primary CNS lymphoma, although an important factor is control of CNS disease immediately prior to transplant.
Tumor lysis syndrome
Tumor lysis syndrome is a common complication after treatment of high-grade, bulky NHLs (due to their exquisite sensitivity to therapy and high proliferative capacity). The syndrome is characterized by renal failure, hyperkalemia, hyperphosphatemia, and hypocalcemia.
Measures to prevent this complication include aggressive hydration; allopurinol; alkalinization of the urine; and frequent monitoring of electrolytes, uric acid, and creatinine. Dialysis is sometimes required. Rasburicase, a recombinant urateoxidase enzyme, is now available for the prevention and treatment of hyperuricemia. (For a more comprehensive discussion of the tumor lysis syndrome, see chapter 39.)
Follow-up of long-term survivors
Relapse The most important risk to patients with NHL is relapse. Among patients with diffuse aggressive lymphomas, most recurrences are seen within the first 2 years after the completion of therapy, although later relapses may occur. Physical examination and laboratory testing at 2- to 3-month intervals and follow-up CT scans (with or without PET scan) at 6-month intervals for the first 2 years following diagnosis are recommended. However, it is recognized that upon relapse of disease, the patient usually presents with symptoms rather than the relapse being diagnosed purely on the basis of surveillance scans or routine clinic visits.
Early detection of recurrent disease is important because these patients may be candidates for potentially curative high-dose therapy and SCT. Patients with advanced low-grade NHL are at a constant risk of relapse, and late recurrence of disease may be seen, sometimes after more than a decade-old remission.
Secondary malignancies Long-term survivors are at increased risk of second cancers. In a survey of 6,171 patients with NHL who survived 2 or more years, nearly 1,000 patients lived 15 or more years after diagnosis. Second cancers were reported in 541 subjects, with significant excesses seen for all solid tumors; acute myeloid leukemia; melanoma; Hodgkin lymphoma; and cancers of the lungs, brain, kidneys, and bladder. The actuarial risk of developing a second malignancy at 3 to 20 years after diagnosis of NHL was 21%, compared with a population-expected cumulative risk of 15%.
Treatment complications There has been a more selective use of irradiation as part of the initial therapy for NHL; therefore, the risk of certain radiation-induced complications has been reduced or eliminated in more recently diagnosed patients. Nevertheless, total-body irradiation may be used as a component of myeloablative conditioning regimens. Also, transplant recipients are at increased risk of secondary myelodysplasia and acute myeloid leukemia, regardless of whether they received a radiation-containing conditioning regimen. All individual chemotherapy agents have their own potential long-term morbidity.
Long-term survivors need continued follow-up for possible treatment-related complications. Some of these toxicities may still be unknown. Careful documentation of late complications will be important in the design of future treatment strategies aimed at preserving or improving response rates and the duration of remission while reducing toxicity.















