Cancer Management Chapter 29: Acute leukemias
Hematopoietic malignancies account for 6% to 8% of new cancers diagnosed annually. In the year 2009, an estimated 44,790 new cases of leukemia were diagnosed, and 21,870 deaths were attributable to leukemias of all types. The total age-adjusted incidence of leukemia, including both acute and chronic forms, is 9.6 per 100,000 population; the incidence of acute lymphoblastic leukemia (ALL) is 1.5 per 100,000 and of acute myelogenous leukemia (AML) is 2.7 per 100,000 population.
Hematopoietic malignancies account for 6% to 8% of new cancers diagnosed annually. In the year 2009, an estimated 44,790 new cases of leukemia were diagnosed, and 21,870 deaths were attributable to leukemias of all types. The total age-adjusted incidence of leukemia, including both acute and chronic forms, is 9.6 per 100,000 population; the incidence of acute lymphoblastic leukemia (ALL) is 1.5 per 100,000 and of acute myelogenous leukemia (AML) is 2.7 per 100,000 population.
Epidemiology
Gender The incidence of both ALL and AML is slightly higher in males than in females.
Age The age-specific incidence of AML is similar to that of other solid tumors in adults, with an exponential rise after age 40. With regard to ALL, 60% of cases are seen in children, with a peak incidence in the first 5 years of life and a subsequent drop in incidence until age 60, when a second peak emerges.
Race and ethnicity The incidence of acute leukemia is slightly higher in populations of European descent. Also, a report from the University of Southern California indicates that acute promyelocytic leukemia (APL) is more common in Hispanic populations than in other ethnic groups.
Etiology and risk factors
There is wide diversity in the behavior of the various subsets of acute leukemias. Thus, it is unlikely that there is one common etiology for these aberrant cellular proliferations. There are, however, some accepted risk factors for leukemogenesis.
Chemical exposure The increased incidence of AML and myelodysplasia (preleukemia) has been reported in persons with prolonged exposure to benzene and petroleum products. The interval between exposure and the onset of leukemia is long (10 to 30 years). Chromosomal damage is common.
Pesticide exposure also has been linked to some forms of AML. The incidence of AML is beginning to rise in developing countries, as industrialization and pollution increase.
Other environmental exposures Exposure to hair dyes, smoking, and nonionic radiation may also increase the risk of leukemia.
Prior chemotherapy or irradiation Use of alkylating agents, such as cyclophosphamide and melphalan (Alkeran), in the treatment of lymphomas, myelomas, and breast and ovarian cancers has been associated with the development of AML, usually within 3 to 5 years of exposure and often preceded by a myelodysplastic phase. Cytogenetic abnormalities, particularly monosomy 5, 7, 11, and 17, are common. Concurrent radiation exposure slightly increases the risk of leukemogenesis posed by alkylating agents.
Topoisomerase II inhibitors (etoposide, teniposide [Vumon], doxorubicin and its derivatives, and mitoxantrone, used to treat ALL, myeloma, testicular cancer, and sarcomas, as well as taxanes used to treat breast cancer, have also been implicated in leukemogenesis. These agents, in contrast to alkylators, are associated with a short latency period without antecedent myelodysplasia and with cytogenetic abnormalities involving chromosome 11q23 or 21q22 in the malignant clone.
Genetic disorders An increased incidence of AML is seen in patients with Down syndrome, Bloom syndrome, or Fanconi's anemia, as well as in individuals with ataxia-telangiectasia or Wiskott-Aldrich syndrome. In identical twins younger than age 10, if one child develops leukemia (usually ALL), there is a 20% chance that the other twin will develop leukemia within a year; subsequently, the risk falls off rapidly and joins that of nonidentical siblings, which is three to five times that of the general population.
Signs and symptoms
Effects on hematopoiesis Leukemia manifests symptomatically by its impact on normal hematopoiesis. Thus, easy fatigability, bruising, and bleeding from mucosal surfaces, fever, and persistent infection are all reflections of the anemia, thrombocytopenia, and decrease in functional neutrophils associated with marrow replacement by malignant cells. Bone pain is common in children with ALL (occurring in 40% to 50%) but is less common in adults (5% to 10%).
Whereas a marked elevation in WBC count is the classic hallmark of leukemia, pancytopenia is more common, particularly in patients of all ages with ALL or in elderly patients with AML, who may have had preexisting marrow dysfunction (myelodysplasia). Only 10% of newly diagnosed patients with either AML or ALL present with leukocyte counts > 100,000/μL. These patients, however, constitute a poor prognostic group and are at increased risk of CNS disease, tumor lysis syndrome, and leukostasis due to impedance of blood flow from intravascular clumping of blasts, which are “stickier” than mature myeloid or lymphoid cells.
Leukostasis may manifest as an alteration in mental status; intermittent or persistent cranial nerve palsies, particularly those involving extraocular muscles; priapism; dyspnea; or pleuritic chest pain, due to small leukemic emboli in the pulmonary vasculature.
Physical findings in AML are usually minimal. Pallor, increased ecchymoses or petechiae, retinal hemorrhage, gingival hypertrophy, and cutaneous involvement are more common with monocytic (M4 or M5) variants of AML than with other variants of AML.
Mild hepatosplenomegaly and lymphadenopathy are seen in many cases, particularly in childhood ALL. Massive hepatosplenomegaly occurs infrequently and should raise the suspicion of a leukemia evolving from a prior hematologic disorder, such as chronic myelogenous leukemia (CML) or myelodysplasia. Mediastinal adenopathy is seen in 80% of cases of T-cell ALL, is less common in other ALLs, and is rare in AML.
Visceral involvement is also rare, occurring as an initial manifestation of AML in < 5% of cases, but it may be more frequent during subsequent relapses. These focal collections of blasts, called chloromas or granulocytic sarcomas, can present as soft-tissue masses, infiltrative lesions of the small bowel and mesentery, or obstructing lesions of the hepatobiliary or genitourinary system.
CNS involvement is uncommon at presentation in adult AML (< 1%) and adult ALL (3% to 5%). In most instances, CNS involvement is detected by screening lumbar puncture in high-risk patients who are asymptomatic at the time of the puncture. Symptoms, when they do occur, include headache, diplopia, cranial nerve palsies, radicular pain, and/or weakness in a particular nerve root distribution. CNS involvement usually is restricted to leptomeninges; parenchymal mass lesions are uncommon.
Like the CNS, the testes appear to be a “sanctuary” for isolated relapses in pediatric but not adult ALL. Signs of testicular involvement include painless, asymmetric enlargement.
Metabolic effects of acute leukemia relate primarily to the rate of cell death.
Hyperuricemia with possible interstitial or ureteral obstruction is seen predominantly in AML with moderate leukocytosis; this condition may be exacerbated by a rapid response to chemotherapy and the “tumor lysis syndrome” (hyperuricemia with renal insufficiency, acidosis, hyperphosphatemia, and hypocalcemia), which may occur within the first 24 to 48 hours after initiating chemotherapy. To prevent this complication, all patients should receive allopurinol and urine alkalinization before marrow-ablative chemotherapy is initiated. In patients with a high tumor burden, renal insufficiency, or acidosis prior to initiation of chemotherapy, rasburicase (Elitek) may offer a more rapid treatment for hyperuricemia.
Coagulopathies can also complicate the hemostatic defects associated with thrombocytopenia. Disseminated intravascular coagulation (DIC) is most often seen in APL (French-American-British Cooperative group [FAB] subtype M3) due to release of procoagulants from the abnormal primary granules, which activate the coagulation cascade, leading to decreased factors II, V, VIII, and X, and fibrinogen, as well as rapid platelet consumption. Lysozyme released from monoblasts in M4 and M5 subtypes of AML can also trigger the clotting cascade. Finally, DIC can occur following L-asparaginase (Elspar) chemotherapy for ALL.
Diagnosis
Abnormalities on the CBC raise the possibility of leukemia. The diagnosis is substantiated pathologically by a bone marrow examination.
All patients should have cytochemistry, immunophenotyping by fluorescent-activated cell sorter (FACS) using monoclonal antibodies directed at leukemia-specific antigens, and cytogenetic analysis of the marrow or peripheral blood blasts at diagnosis. Other tests used to evaluate metabolic abnormalities (electrolytes, creatinine, and liver function tests) and coagulopathies are also needed at diagnosis. A lumbar puncture should be performed at diagnosis in all pediatric patients with ALL and in all patients with neurologic symptoms regardless of age and pathology.
Pathology and cytogenetics
Acute leukemias comprise a group of clonal disorders of maturation at an early phase of hematopoietic differentiation. Morphology and cytochemical stains designed to detect intracellular myeloperoxidase or esterases have been the traditional methods used to classify acute leukemias into either myeloid or lymphoid derivations.
Coupling these traditional methods with cytogenetic analysis and highly specific monoclonal antibodies directed against cell-surface antigens has led to the detection of new prognostic factors and has provided an approach to detect minimal residual disease.
In 1997, a panel of hematopathologists met to update the FAB classification of hematologic



malignancies, which was based on morphology and cytochemistry alone. They proposed a new classification, incorporating immunophenotyping, cytogenetics, and clinical disease features, which has been adopted by the World Health Organization (WHO; Tables 1 and 2).
Myeloid leukemias
The WHO classification retains the morphologic subgroups of the FAB system in the subgroup of “AML not otherwise categorized” but has created new categories that recognize the importance of certain cytogenetic translocations as predictors of response to therapy. In this category are AML with t(8;21)(q22;q22), AML with abnormal eosinophils and inv(16)(p13;q22) or t(16;16)(p13;q11), AML with 11q23 mixed-lineage leukemia abnormalities, and APL with t(15;17)(q22;q11-12) or variants (Table 1).



The WHO classification also attempts to deal with the evidence that, in many older patients, marrow dysfunction antedates the onset of acute leukemia. These myelodysplastic syndromes (MDSs) are characterized by ineffective hematopoietic production and disrupted maturation of one or more cell lines. These abnormalities are often accompanied by loss of chromosomal material, particularly -5 or -5q, -7 or -7q, and -3 or -20. As the bone marrow becomes more dysfunctional, increasing numbers of blasts are seen in the marrow.
In the FAB classification, the demarcation line between myelodysplasia and AML was 30% marrow blasts. However, patients with 20% to 29% blasts (previously classified as refractory anemia with excess blasts in transition [RAEB-t]) have a biologic behavior and poor survival similar to those of patients with AML. WHO lowered the threshold for the diagnosis of AML to 20% marrow blasts and deleted the FAB category of RAEB-t. In addition, patients with 5% to 20% blasts who have t(15;17), t(18;21), or inv(16) are considered to have AML rather than MDS and should receive AML treatment.
The WHO system further subdivided the AML patients with dysplastic maturation into those with or without antecedent cytopenias (usually 3 months prior to diagnosis had been the arbitrary cutoff point) and those with a history of exposure to chemotherapy agents (alkylating agents, epipodophyllotoxins, or others). In 2003, the International Working Group for the Diagnosis and Standardization of Response Criteria accepted the WHO classification as the standard for AML diagnosis.
The genetic profile of malignant cells has been found to vary widely from normal, with many genes being either overexpressed or suppressed. DNA microarray techniques allow the simultaneous analysis of thousands of genes that are being studied in AML and ALL for their predictive ability to define cohorts of patients with similar outcomes; this process may in turn allow the selection of candidate genes that can be used as therapeutic targets in the future.
Lymphoblastic leukemias
Lymphoblastic leukemias can arise from either B-cell or T-cell progenitors that arrest at an early stage of maturation and then proliferate. Marrow involvement of > 25% lymphoblasts is used as the demarcation line between lymphoblastic lymphoma, in which the preponderance of tumor bulk is in nodal structures, and ALL. Approximately 75% of adult ALLs are B cell in derivation and 25% are T cell.
Precursor B-cell ALL Most B-cell leukemias are early or “pre-B” cell, expressing CD19 and CD10 (the common acute leukemia antigen [cALLa]) but lacking surface or cytoplasmic immunoglobulin; this group of early B-cell leukemias has a more favorable prognosis than that of B-cell leukemias in which the cells have a more mature phenotype. Chromosomal rearrangements juxtaposing an oncogene with a promoter region are often seen in this disease category (Table 2).
<>Mature B-cell ALL The more mature B-cell ALL, or Burkitt-cell leukemia, is associated with translocations of the c-myc gene on chromosome 8 and the immunoglobulin heavy-chain gene on chromosome 14q32 in 80% of cases or with the light-chain genes of chromosome 2p11 or 22q11 in the other 20%. Burkitt-cell leukemia has increased in frequency recently, as it is one of the lymphoproliferative disorders that occur in individuals infected with the human immunodeficiency virus (HIV); leukemia may appear early in the course of the HIV infection, before the onset of opportunistic infections or severe T-cell deficiency (see chapter 24).
A small fraction (2%) of patients with precursor B-cell ALL lack CD10 expression. Patients with CD10-negative disease have a high incidence of MLL gene expression (83%) and a very poor disease-free survival (DFS; 12%) at 2 years.
T-cell ALL is frequently associated with translocations of T-cell receptor genes on chromosome 14q11 or 7q34 with other gene partners. T-cell ALL had been associated with a poor prognosis when treated with conventional ALL regimens but now is associated with a better prognosis if treated with aggressive antimetabolite therapy. Precursor T-cell ALL has a poorer outcome.
Infection with human T-cell leukemia virus-1 (HTLV-1) should be looked for in patients with T-cell ALL presenting with hypercalcemia and lytic bone lesions. HTLV-1 infection is endemic in southern Japan, the southern Pacific basin, the Caribbean basin, and sub-Saharan Africa. High infection rates are also seen in parts of Iran, India, and Hawaii. Recent immigrants from endemic areas retain a risk of infection similar to that of their point of origin. However, fewer than 0.1% of persons carrying HTLV-1 will develop T-cell leukemia.
ALL with myeloid antigen expression vs undifferentiated leukemia
A subset of patients with leukemia exhibits features of both myeloid and lymphoid differentiation. These patients were originally classified as having mixed-lineage leukemia. Patients with a leukemic clone that expresses two or more ALL antigens and one myeloid antigen comprise 20% of adult ALL cases. Although expression of myeloid antigen is considered to be a poor-risk feature in children, it does not constitute a distinct poor-risk feature in adults.
Immunophenotyping has also helped define a group of patients with undifferentiated myeloid leukemia (M0) who previously were likely to be treated as if they had ALL. These leukemias have a primitive morphology and lack myeloperoxidase. On immunophenotyping, they express at least one early myeloid antigen, usually CD13 or CD33, and no T- or B-cell markers. Based on immunophenotyping, undifferentiated leukemias are now recommended to be treated in the same manner as myeloid malignancies.
ALL prognostic factors
Factors known to have an impact on the ability to achieve and maintain remission in ALL include age, lineage derivation, elevated WBC count, and cytogenetic abnormality. The cure rate for children aged 2 to 12 is over 80%; this 5-year DFS rate drops to 40% to 60% for younger adults and is less than 30% for adults over age 65. Poorer outcomes in adults have been attributed to a higher proportion of patients with poor-risk karyotypes, particularly t(9;22) or the Philadelphia chromosome(Ph), as well as poorer tolerance to chemotherapy, especially L-asparaginase and high-dose methotrexate.
The recent 1,500 patient MRC XII/ECOG adult ALL trial reconfirmed that age (> 35 years), elevated WBC count, B-cell lineage, and karyotypic abnormalities are significant independent risk factors for DFS and overall survival (OS). In addition to the previously reported poor-risk karyotypes of t(9;22), t(4;11), and t(8;14), this trial also identified complex (≥ 5) abnormalities and low hypodiploid/near triploid as new poor-risk features.
Philadelphia chromosome The most common cytogenetic abnormality in ALL is the translocation of the abl gene from chromosome 9 to the breakpoint cluster region on chromosome 22, forming a new gene product (BCR-ABL) with tyrosine kinase activity. This translocation, referred to as the Philadelphia chromosome (Ph), is found in 95% of cases of CML and in 20% to 30% of newly diagnosed adults with ALL.
The fusion protein produced by the BCR-ABL translocation in Ph+ ALL (p190) differs from the product seen in CML (p210); the p190 product is a smaller protein than the p210 product and has higher tyrosine kinase activity. Use of polymerase chain reaction (PCR) techniques that target only the p210 product will significantly underestimate the incidence of Ph+ ALL. In a recent update of the German ALL trials, 37% of patients were Ph+, with 77% showing the p190 product vs 23% showing the p210 product.
Although patients with Ph+ ALL may attain a morphologic remission with conventional chemotherapy (82%), almost all such patients will have persistent molecular evidence of disease on PCR. Patients who do achieve a molecular remission have a longer duration of remission than those who continue to express p190 or p210 activity (30 vs 12 months).
Recently, GIMEMA, the Italian oncologic cooperative group, published outcomes data of a large trial of adult patients with ALL in which both cytogenetic data and molecular probes for specific gene products were combined to define prognostic groups. The molecular abnormalities that were evaluated were t(9;22) BCR-ABL, t(4;11)/ MLL-AFA, t(1;19) E2A/PBX1, 9p/p15-p16 deletions, and 6q deletions. Categories based primarily on classic karyotypes were normal, hyperdiploid, and miscellaneous structural abnormalities of uncertain significance.
The use of molecular probes was particularly informative in patients with failed karyotypic analysis or normal cytogenetics. The use of the BCR-ABL probe increased the number of cases with a t(9;22) abnormality from 64 to 104 (26% of patients in the trial); more than 50% of add(9p)/p15-p16 abnormalities were detected only by molecular testing. Patients with t(9;22), t(4;11), and t(1;19) had disease-free intervals of 0.4 to 0.6 years, whereas those with del(6q), hyperdiploid, or pseudodiploid karyotypes had intermediate DFS of 1.3 to 1.6 years; those with a normal karyotype or del(9p)/p15-p16 had better outcomes (2.9 and 4 years, respectively).
Other translocations Translocations involving the mixed-lineage leukemia gene at chromosome 11q23 are partnered with several other chromosomes, including 4q21, 9q22, and 19q13. Translocations involving chromosome 11q23 are frequently seen in secondary leukemias, particularly those arising after chemotherapy with etoposide or teniposide. Although most of these translocations are associated with AML, ALL has also arisen in this setting. All the 11q23 translocations, as well as the more common (1;19) translocation, are associated with poorer outcomes when compared with similar immunophenotypes coupled with normal cytogenetics.
Molecular techniques, such as PCR amplification of leukemia-specific sequences of RNA or DNA, have been used in research settings to reveal residual leukemia cells. These sensitive techniques can detect the persistence of cells with the leukemic phenotype at a sensitivity of 1 cell in 104 normal cells in patients who are deemed to be in complete remission (CR) by conventional techniques. In two pediatric studies, detection of leukemia-specific gene rearrangements ( ≥ 1 cell in 104 normal cells) 5 to 6 months after initiation of treatment was associated with a high relapse rate.
A confirmatory study in adults with standard-risk ALL using a combination of aberrant immunophenotyping and PCR amplification of T-cell receptor or immunoglobulin gene rearrangements was recently reported. Patients who had no detectable residual disease by day 11 from the start of induction therapy had a 3-year DFS rate of 92%, compared with 65% for those with no residual disease by week 16. Patients who had detectable minimal residual disease beyond that point had a 3-year DFS of only 12%.
Time to remission and measurement of minimal residual disease have also been identified as prognostic factors in several pediatric trials. Molecular techniques such as PCR amplification of leukemia-specific sequences of RNA or DNA have been used to detect persistence of cells with the leukemic phenotype at a sensitivity of 1 cell in 104 normal cells. In two pediatric studies, the detection of 7 1 × 10-4 cells at 6 months post diagnosis was associated with a very high relapse rate.
Treatment strategy
Treatment for patients with ALL and AML can be subdivided into two or three phases. Induction chemotherapy is the initial treatment designed to clear the marrow of overt leukemia. This phase usually involves multiple drugs that cause pancytopenia for 2 to 3 weeks.
The purpose of consolidation therapy is to further reduce the residual leukemic burden in patients who are in morphologic remission. Molecular markers of residual disease can often be detected after induction chemotherapy, which indicates the need for further treatment. The intensity of consolidation therapy varies, depending on the risk of relapse (based primarily on cytogenetic or molecular risk groups) and patient age.
Maintenance chemotherapy using low-dose oral chemotherapy for 18 to 24 months has been shown to prolong relapse-free survival in pediatric patients with ALL and in adults with APL. Its value is less clear in adults with ALL; maintenance is used much less frequently in AML.
Treatment of ALL
Induction therapy
The initial goal of therapy is to rapidly reduce the leukemic burden to a level undetectable by conventional methods of light microscopy and flow cytometry, a state that is deemed a CR. Two standard induction regimens have been used in adults with ALL-the Hoelzer regimen, developed by the BFM multicenter group, and the Larson regimen, developed by the CALGB. Along with the standard induction schemas, two newer regimens, the Hyper-CVAD (high-dose cyclophosphamide, vincristine, adriamycin, adriamycin [doxorubicin], dexamethasone) regimen from M. D. Anderson and the Linker regimen (2002 version), which have a similar induction drug dosing as the older regimens but include much higher doses of antimetabolites (cytarabine [Ara-C] and methotrexate) and etoposide for dose-dense consolidations, are outlined in Table 3 along with the standard induction schemas. Overall, CRs are obtained in 80% to 94% of adults younger than age 60 treated with any of these regimens. The incidence of death during induction therapy for these trials was low (2% to 9%).
The addition of an anthracycline to the standard pediatric leukemia induction regimen of vincristine, prednisone, and L-asparaginase increased the CR rate in adults from 50% to 60% to 70% to 85% in several series. In a recent CALGB study, the use of cytokines, ie, granulocyte colony-stimulating factor (G-CSF, filgrastim [Neupogen]), during induction therapy in patients older than 60 years of age, reduced treatment-related mortality from 31% to 5% when compared with placebo-treated controls.
L-asparaginase has been a major agent in pediatric trials in both induction and consolidation therapies. Although L-asparaginase is used during induction therapy in adults younger than age 50 with ALL (except in the M. D. Anderson Hyper-CVAD regimen), it is rarely used in consolidation therapy. The potential importance of this drug was emphasized by observations on L-asparaginase depletion during induction therapy on the most recent CALGB trial, which showed a median survival of 31 months in patients who were depleted vs 13 months for those who were not depleted. They also showed improved depletion using the pegylated form of L-asparaginase (pegaspargase [Oncaspar]), which has a longer half-life.
Assessment of outcomes for young adults (< 30 years) treated on either pediatric or adult regimens has shown comparable remission rates but significant differences in long-term disease-free survival. As a rule, the pediatric regimen contains higher and more frequent doses of L-asparaginase as well as stricter adherence to a tight dose schedule. All three US adult cooperative groups are now collaborating on trial with the COG using one arm of the high-risk ALL protocol for patients < 30 years old to see whether comparable results can be obtained.
A recent Canadian study using a modified pediatric regimen with weekly high-dose L-asparaginase for 30 weeks during intensification in adults aged 18 to 60 years showed an overall survival of 63% and a disease-free survival of 71% at 5 years. Adverse predictors of outcome included age > 35 years, MLL gene rearrangement, high WBC count, and < 80% planned L-asparaginase dose. The regimen was associated with significant morbidity, however, including infections (47%), avascular necrosis of major joints (32%), thromboembolic events (23%), and peripheral neuropathy (22%).
The FDA has approved pegaspargase (Oncaspar) as a component of a multiagent chemotherapy regimen for the first-line treatment of patients with ALL. The drug had originally been approved only for ALL patients who were allergic to native forms of L-asparaginase. A single dose of 2,000 IU/m2 is now used to replace the multiple doses on the Linker regimen as well as CALGB induction and intensification (Table 3).
T-cell ALL There is evidence that patients with T-cell ALL may benefit from early treatment with Ara-C and cyclophosphamide. Pharmacologic studies show high levels of Ara-C triphosphate accumulation in T lymphoblasts and synergy between cyclophosphamide and Ara-C in cell lines of T-cell malignancies. T lymphocytes also have a lower expression of polyglutamate synthetase than pre-B blasts. Randomized trials in children with T-cell ALL showed that the use of high-dose methotrexate (up to 5 g/m2) also improved outcome.
Mature B-cell ALL Patients with the more mature B-cell ALL (Burkitt-cell leukemia) experienced an improvement in survival when high doses of cyclophosphamide, methotrexate, and Ara-C were incorporated early in the treatment course. The probability of leukemia-free survival improved from 35% with standard ALL induction to 60% to 70% with these newer regimens. Rituximab (Rituxan) is also being added in patients whose blasts express CD20.
Ph+ ALL
The development of imatinib (Gleevec), a selective BCR-ABL protein kinase inhibitor, provided a potent new agent in the treatment of Ph+ ALL. As a single agent, it produced CRs in 30% of patients with relapsed Ph+ disease. Several centers have reported improved remission rates of 90% to 95% when imatinib (600 mg/day) was added to initial induction therapy (usually in regimens lacking L-asparaginase) without additional toxicity. With continuation of imatinib through consolidation therapy, 50% to 60% of patients will achieve a molecular remission by 60 days post induction. Patients who achieve a molecular remission and continue on imatinib therapy have an improved disease-free survival of 62% at 3 years, compared with 14% at 1 year in the pre-imatinib era.
Consolidation therapy
The BFM, CALGB, Linker (2002), Hyper-CVAD, and COG consolidation regimens for ALL are outlined in Table 3. There is a CALGB Intergroup Study regimen for adults age 30 or younger which uses more dose-intensive consolidation with asparaginase usage (for more information about this, visit www.ClinicalTrials.gov and search for trial NCT 00558519). As yet, no randomized trials have compared these regimens. However, in sequential studies from Memorial Sloan-Kettering Cancer Center, BFM group, and the Linker study, use of multiple cycles of non-cross-resistant drugs for 3 to 8 cycles after remission followed by maintenance with methotrexate and mercaptopurine (Purinethol) resulted in overall long-term DFS rates of 38% to 52%.
Long-term outcome data of 288 patients treated with Hyper-CVAD showed an 81%



CR rate after cycle 1A and a 92% rate after receiving both cycles 1A and 1B (see Table 3); a 5% overall death rate during induction was noted, although treatment-related mortality reached 15% in patients older than age 60 despite the use of G-CSF (granuolocyte colony-stimulating factor). At a median follow-up of 63 months, the 5-year DFS was 38%, similar to that reported in the BFM and CALGB trials. In this series, adverse prognostic factors for DFS were age ≥ 45 years, poor performance status, WBC count > 50,000/μL, Ph+ cytogenetics, more than 1 cycle to achieve a CR, or > 5% residual blasts at day 14. Patients with none or one of these factors had a 52% 5-year DFS rate, vs 37% for patients with two or three factors and only 10% for patients with at least four risk factors.
In the 2002 Linker trial, which intensifies the consolidation with alternating cycles of higher-dose Ara-C (HDAC) and etoposide alternating with cycles of high-dose methotrexate, the 5-year relapse-free survival rate was 52% overall and 60% for patients with standard-risk features. Prognostic features that were associated with a poor outcome in this study included pre-B ALL with > 100,000/μL WBC count at diagnosis, cytogenetic abnormalities involving chromosome 11q23 or t(9;22), and time to remission > 30 days. Without either allogeneic or autologous transplantation, all high-risk patients relapsed within a short time (1 to 9 months).
The French LALA-94 trial of 922 patients was designed to look at postremission therapy that was stratified by risk of relapse. The standard-risk patients who achieved CR with 1 cycle of induction therapy were randomized to receive either conventional cyclophosphamide, Ara-C, and mercaptopurine or early intensification with intermediate-dose Ara-C (1 g/m2 × 8 doses) and mitoxantrone.
In this study, there was no difference in 5-year DFS (33% conventional vs 37% early intensification, with an OS at 5 years of 44%). High-risk patients included those with defined cytogenetic risks (excluding Ph+), WBC count > 30,000/μL, and CNS disease at diagnosis or who required more than 35 days to achieve CR. Patients with a sibling donor received allogeneic transplant in CR, with the remainder randomized to receive either the early intensification chemotherapy or autologous transplant. The 5-year DFS was 45% for those receiving allogeneic transplant and 23% for those without a donor. There was no significant difference in OS with chemotherapy vs autologous transplant, but there was a different pattern of relapse, with fewer late relapses in the autologous patients.
High-risk patients Although the BFM regimen is now standard therapy for standard-risk patients (aged 30 to 55 years), high-risk patients are being selected for dose-intensive therapies, including HDAC and methotrexate or etoposide, high-dose methotrexate, and L-asparaginase.
The addition of imatinib during induction therapy has resulted in a CR rate of 95% to 100% after 1 cycle of induction, and patients have achieved molecular remission documented by PCR in 50% to 60% of patients within 2 months. Introduction of imatinib early during induction and consolidation has significantly improved outcomes for this high-risk group. Patients with high-risk features including Ph+ have DFS of 50%-60% when allogeneic stem cell transplant is performed in CR for adults.
Allogeneic hematopoietic transplantation (HCT)
Myeloablative allogeneic HCT combines dose-intensive chemotherapy and radiation therapy with the immunotherapeutic aspects of graft-vs-leukemia effect from donor antitumor surveillance. Relapse rates following allogeneic HCT for high-risk patients are significantly lower (15% to 20% vs 50% to 65%) for nontransplant recipients, but transplant-related mortality is also high (20% to 30%) and increases with age. Several studies have shown improved survival for high-risk patients receiving allogeneic transplants in first CR. Results from the ECOG/MRC trial also showed improved disease-free survival (62% vs 52%) at 5 years for standard-risk patients transplanted in first CR. Three sequential French ALL trials as well as the ECOG/MRC trial failed to show any survival advantage for autologous HCT compared with 2.5 to 3 years of consolidation and maintenance chemotherapy.
For patients with Ph+ ALL, imatinib has provided a means to achieve molecular remission in approximately half of these very high-risk patients; this has allowed physicians more time to identify an unrelated donor. Studies that will assess the impact of molecular remission pre-HCT on the risk of relapse are in progress. Imatinib (at 400 mg/day orally) is also studied in the post-HCT phase. Patients who remain molecularly positive or revert to a positive state are at high risk of relapse and should be considered for treatment with second-generation tyrosine kinase inhibitors such as dasatinib (Sprycel) or nilotinib (Tasigna). (See CML chapter for further detail).
CNS prophylaxis
CNS relapse occurs at a much higher frequency in patients with ALL than in those with AML. The rate of CNS relapse was 20% in the first year in a pediatric ALL trial in which the CNS therapy was attenuated to a subtherapeutic level.
Patients with ALL require preemptive therapy for occult CNS disease with either (1) intrathecal methotrexate and/or Ara-C combined with cranial irradiation or (2) high-dose systemic Ara-C or methotrexate combined with intrathecal therapy. Specific use of intrathecal liposomal Ara-C should not be used concomitantly with high-dose systemic chemotherapy such as Ara-C, methotrexate, or etoposide, which cross the blood-brain barrier due to a high risk (15% to 20%) of serious neurotoxicity (seizures, cauda equina syndrome, and encephalitis).
Maintenance therapy
Maintenance therapy with daily mercaptopurine and weekly methotrexate for 18 to 24 months beyond consolidation remains the standard of care for children with ALL. In adults, the benefit of maintenance therapy is less certain. In low-risk adults, who may have an outcome more similar to that in the pediatric population, maintenance therapy would appear to be justified (see Table 3 for maintenance regimens). In individuals who have mature B-cell ALL, it is unlikely that maintenance therapy has any effect. In other high-risk adult populations, more than half of patients relapse while on maintenance therapy, indicating the need for other strategies to eradicate minimal residual disease.
Treatment of relapse
Treatment of relapsed adult ALL is a major challenge. Because most protocols for initial treatment incorporate 6 to 11 agents with different cytotoxic mechanisms, a selection process for drug resistance has occurred. The overall remission rate for relapse therapy is 30% to 40%, with a median duration of remission of 6 months. In the MRC/ECOG trial, the 5-year overall survival for adults who relapsed was 7% in the absence of allogeneic transplant.
Salvage strategies include reinduction with the initial regimen in patients with late relapse or high-dose antimetabolites (Ara-C or methotrexate [see Hyper-CVAD regimen, Table 3]) in those who relapse early. Recent experimental approaches include monoclonal antibodies directed against leukemia-specific antigens conjugated to either radionuclides or toxins, tyrosine kinase inhibitors, allogeneic or autologous transplantation, or new agents.
Clofarabine (Clolar) has been approved for treatment of relapsed refractory ALL in children. Of 61 patients, 12 achieved CR, including children who had relapsed following allogeneic transplantation. The maximum tolerated dose was 52 mg/m2 infused over 2 hours daily for 5 days. Significant toxic effects include febrile neutropenia, anorexia and nausea, capillary leak syndrome, hepatotoxicity, and skin rash.
In individuals with Ph+ ALL or CML in lymphoid blast crisis, imatinib (400 to 800 mg orally daily) can induce remissions in up to 30% of patients. These remissions are short-lived but may control the leukemia long enough for a donor to be identified, thus providing an option for an allogeneic transplant in second remission. Nilotinib, which has been recently approved by the FDA, is an imatinib analog with high binding affinity to BCR-ABL. It has been shown to overcome imatinib resistance in approximately 30% of Ph+ ALL patients.
Dasatinib, which is a kinase inhibitor of multiple targets including BCR-ABL, c-Kit, SRC, and PDGFR (platelet-derived growth factor receptor) can provide short-term salvage therapy for patients whose disease progresses while receiving combinations of imatinib and chemotherapy. Dasatinib has a higher propensity for complications related to serositis, with significant pleural and peritoneal effusion. There is some evidence of CNS penetration for dasatinib compared with imatinib in patients with Ph+ ALL who develop active CNS disease. They should be switched to dasatinib in conjunction with intrathecal chemotherapy.
Nelarabine (Arranon) has been approved for the treatment of T-cell lymphoblastic disease. In a recommended dose of 1,500 mg/m2 on days 1, 3, and 5, this agent has produced response rates of 30% to 50% in heavily pretreated patients. Vinorelbine has produced remission in 50% of adults with relapsed ALL in a small pilot study.
TREATMENT OF AML
Although the chemotherapeutic agents used in the initial therapy for AML have not changed much in the past 30 years, our knowledge of the biology of leukemia has increased. The identification of prognostic factors can provide more realistic expectations of response to standard treatment and can define the population for whom investigational therapy is appropriate early in the course of disease.
Prognostic factors Cytogenetic abnormalities are the major predictors of remission and risk of relapse for patients with AML. Patients with translocation of genetic material involving core binding regions [t(15;17), t(8;21) inv(16), or t(16;16)] have a good prognosis, with remission rates of 88% and 5-year DFS rates of 55% to 80%, whereas patients with loss of genetic material from chromosome 5 or 7 (-5 or -5q, -7 or -7q) and complex karyotypic abnormalities (defined as more than five abnormalities) have lower rates of CR (30% to 40%) and DFS (5%) at 5 years. Patients with either normal or intermediate cytogenetic abnormalities have a CR rate of 67% and a 5-year DFS rate of 25% to 30%, based on data from a large CALGB trial using HDAC-based consolidation therapy.
Internal duplication of FLT3 can be found in one-third of patients with normal cytogenetics or in patients with t(15;17) (APL) but is uncommon in either poor-risk karyotypes or non-APL translocations. This abnormality does not appear to have an impact on remission, but it is a predictor for relapse (74% relapse rate in patients with a normal karyotype with an isolated FLT3 mutation vs 46% for patients with wild-type FLT3). In patients with otherwise favorable cytogenetic abnormalities [t(8;21) or inv(16)], the presence of c-Kit mutation increases the risk of relapse.
In a phase I/II dose-finding trial of clofarabine (Clolar) combined with cyclophosphamide and etoposide, a 55% complete remission or complete pathologic response was achieved among 25 patients with relapsed/refractory ALL, with a median remission duration of 34 weeks. However, four patients developed severe liver toxicity, including veno- occlusive disease in patients with prior SCT or with hepatitis (Hijiya N et al: Blood 112: abstract 2925, 2008).
Mutations of nucleophosmin protein (NPM-1), which shuttles nucleic acids and proteins from the nucleus to the cytoplasm as well as binding p53, are also a commonly reported abnormality, prevalent (47%) in patients with a normal karyotype. Although there is frequent overlap with FLT3 mutations, patients with an isolated NPM-1 mutation and a normal karyotype have a 60% DFS vs 40% for those with either wild-type or mutations of both FLT3 and NPM1 and 20% for those with an isolated FLT3 mutation. Other molecular mutations in patients with normal cytogenetics that have been reported to favorably impact relapse-free survival are CEPBA and NRAs, while MLL partial tandem duplication carries an unfavorable implication.
Poor-risk cytogenetics, antecedent MDS, and a high incidence of multidrug resistance (MDR-1) protein are found more commonly in patients older than age 60, which accounts for the lower CR rates (30% to 55%) seen in older individuals compared with their younger counterparts (60% to 80%). Many older patients with preexisting MDS may clear marrow blasts with antileukemic treatment but may still have impaired hematopoiesis and persistent cytopenias, since they may have no residual normal stem cells to repopulate the marrow.
Induction therapy
Ara-C and an anthracycline such as daunorubicin or idarubicin have been the stand


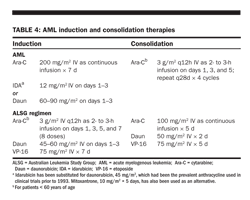
ard drugs used for AML induction chemotherapy for 30 years (Table 4). Depending on the prognostic groups, remission rates of 60% to 80% are seen in younger (< age 60) patients and of 35% to 55% in patients older than age 60. Other agents such as mitoxantrone and etoposide also have antileukemic activity, but no significant increase in remission rates or relapse-free survival has been seen when mitoxantrone was substituted for an anthracycline or etoposide was added to infusional Ara-C and daunorubicin.
Mitoxantrone and etoposide were compared with Ara-C and daunorubicin as induction for patients older than age 55 in SWOG trials; CR rates were 44% for Ara-C and daunorubicin and 33% for mitoxantrone and etoposide, and median survival was 8 and 6 months, respectively. Older patients (> 65 years) with poor-risk cytogenetics have a CR rate of < 30% with standard induction chemotherapy, whereas those with a normal or favorable karyotype have a CR rate of 45% to 50%. Therefore, in older patients, deferring treatment until prognostic cytogenetic information is available may identify patients who will benefit from novel approaches.
One strategy to improve remission rates in younger patients has been to use higher doses of Ara-C during induction therapy. Both the ALSG and the SWOG compared standard Ara-C and daunorubicin (and etoposide in the ALSG trial) with HDAC in patients < 50 years (Table 4). The CR rates were 71% and 74% for standard vs high-dose therapy in the ALSG study and 55% vs 58% in the SWOG trial. In both studies, there was a significantly higher DFS for the high-dose arm at 5 years (48% vs 25% for ALSG and 33% vs 22% for SWOG) but no difference in OS due to increased early toxicity.
At ASCO 2008, ECOG reported the outcome of a phase III trial of daunorubicin (90 mg/m
2
on days 1–3 vs 45 mg/m
2
on days 1–3 during induction therapy) for 633 patients younger than age 60. Complete remission rates were 63% for the higher dose vs 48% for the lower dose (
P
= .0003), with a 5% rate of early death. Median survival was superior at 23.7 months for high-dose daunorubicin vs 15 months for the standard dose (
P
= .003). Older patients given 12 mg/m
2
IV idarubicin × 3 days had more rapid clearance of marrow blasts than patients aged 50 to 70 given daunorubicin at doses of up to 80 mg/m
2
. Given these two trials the optimal anthracycline dose for patients < 65 should be considered to be either idarubicin 12 mg/m
2
× 3 days or daunorubicin 60–90 mg/m
2
IV × 3 days. Higher dose daunorubicin did not benefit patients > 65. A SWOG trial had previously shown 60 mg/m
2
of daunorubicin to be superior to 45 mg/m
2
(Fernandez HF et al: N Engl J Med 361:1249–1259, 2009)
.
Subgroups of patients may benefit from HDAC. In the SWOG trial, patients with CD34+ blasts had a low CR rate of 36% with standard Ara-C but an equivalent rate in those with CD34- blasts (58%) when treated with HDAC. There was a strong correlation between CD34 positivity and MDR-1 expression in this cohort, leading to the inference that HDAC might help overcome drug resistance.
However, a 1,700-patient German trial showed no difference in DFS when 2 cycles of HDAC and mitoxantrone (HAM) were compared with 1 cycle of the standard Ara-C-containing regimen followed by HAM. The overall DFS was 40% for both arms in patients younger than age 60 and 29% for those older than age 60; 80% of young patients received both cycles, whereas only one-third of patients over 60 received cycle 2 irrespective of dose intensity of the initial cycle.
Gemtuzumab ozogamicin (Mylotarg), an anti-CD33 antibody conjugated to the drug calicheamicin, was originally approved for the treatment of relapsed AML in older patients. Currently, there are clinical trials in Great Britain, Italy, and the United States evaluating the addition of gemtuzumab ozogamicin to standard Ara-C and daunorubicin or fludarabine, idarubicin, Ara-C, and G-CSF as initial therapy in patients younger than age 60. Preliminary reports from the British trial showed a CR rate of 86% in patients < 60 years old.
Therapy-related AML has a particularly poor prognosis. At best, only 50% of patients will achieve a remission, usually of brief duration (median, 5 months), despite the use of aggressive drug combinations. Allogeneic or unrelated-donor transplants appear to offer the only curative option in these patients, achieving a 3-year DFS rate of 25% in two studies of allogeneic transplantation.
Initial Treatment of AML in Older Patients
The majority of AML patients are older than age 60 and constitute a group whose disease has a higher prevalence of unfavorable cytogenetics. Many also have poor marrow regenerative characteristics due to prior myelodysplasia. These patients are frequently burdened with comorbid conditions, which make them poor candidates for conventional Ara-C/anthracycline-based chemotherapy. Cytogenetics and performance status were the two most important predictive factors in this age group, followed by age in 5-year increments and secondary AML.
Thus, before initiating chemotherapy for older patients, it may be helpful to await the results of cytogenetic and molecular mutational profiles before choosing conventional chemotherapy in those “fit” for such therapy. Recent trials from the GLSG showed a benefit to chemotherapy in patients older than age 60 using Ara-C, idarubicin, and etoposide in those with either favorable cytogenetics or normal karyotypes with NPM-1-positive/FLT3-negative mutational status, with a complete remission rate over 60% and a remission duration of 1,125 days. In other subgroups, the remission rates were 42%, with a remission duration of 440 days, and only 210 days for those who were FLT3-positive. Given these underwhelming outcomes combined with 30-day mortality in excess of 15% in many trials of Ara-C-based chemotherapy, new agents are being assessed as initial therapy for patients with abnormal karyotypes.
Given that between one-third and one-fourth of older patients have a history of cytopenias or chemotherapy for a prior malignancy, trials of hypomethylation agents such as azacitidine (Vidaza) or decitabine (Dacogen) have been reasonable starting places. Trials of decitabine (20 mg/m2 IV over 1 hour daily for 5 days) reported a CR rate of 26% when used alone or 49% when given at the same dose for 10 days combined with valproic acid. Clinical trials combining decitabine with a histone deacetylating agent, such as vorinostat (Zolinza), are now in progress.
Another novel combination recently reported was azacitidine (75 mg/m2 SC for 7 days) combined with gemtuzumab ozogamicin (3 mg/m2 on day 8). This phase II trial reported a CR rate of 70% (14 of 20 patients), with only a 5% 30-day mortality, compared with a CR rate of 26% for gemtuzumab ozogamicin as a single agent (at a dose of 9 mg/m2) in a similar population, with a mortality rate of 15%. However, the remission duration was short, at a median of 8 months, for the combination, with a median survival of only 10 months.
Clofarabine is also being evaluated as a single agent for older patients with AML. In phase II trials using a dose of 30 mg/m2 IV for 5 days for induction therapy, CR was achieved in 38% of patients older than age 60 (median, 71 years). Patients achieving remission received a maximum of 6 cycles of consolidation therapy (at 20 mg/m2 for 5 days). The median DFS was 34 weeks, with a median survival of 59 weeks for those achieving CR or complete pathologic response. The 30-day mortality was 13% for those older than age 70. At the present time, the FDA has not approved the use of clofarabine for older AML patients pending the outcome of ongoing phase III comparative induction trials.
Gemtuzumab ozogamicin has also been used as a single agent for induction in patients > 61 years who were considered too frail for conventional therapy with Ara-C and daunorubicin. A CR rate of 26% was achieved in 7 patients aged 61 to 75 years; no benefit was seen in 22 patients older than age 75 due to high early mortality (7 of 22 patients).
Farnesyl transferase inhibitors such as tipifarnib and tandutinib are being evaluated as initial therapy for patients over age 70 with newly diagnosed AML. The CR rate for tipifarnib as a single agent was 15% in several phase I/II trials. As tipifarnib did not have significant enough activity to use alone for induction therapy, it is currently being evaluated as maintenance therapy post standard induction chemotherapy.
Consolidation therapy
Once remission of AML is attained, consolidation chemotherapy is required to achieve a durable remission or cure. Standard consolidation regimens are listed in Table 4.
In a CALGB study, 596 patients in CR were assigned to receive 4 courses of postremission Ara-C in one of three dosages: 100 mg/m2 as a continuous infusion for 5 days, 400 mg/m2 as a continuous infusion for 5 days, or 3 g/m2 as a 3-hour infusion every 12 hours on days 1, 3, and 5. For patients ≤ 60 years old, the percentage of patients in CR at 4 years was significantly higher in the HDAC group (44%) than in either the 400-mg/m2 or 100-mg/m2 group (29% and 24%, respectively). For patients > 60 years old, consolidation dose intensity had no impact on DFS, with all groups plateauing at a rate of 16% by 2 years.
Other approaches to consolidation therapy include 1 to 3 cycles of consolidation followed by autologous or allogeneic bone marrow transplantation. Both of these approaches also tend to be limited to patients < 60 years old and have produced long-term DFS rates of 45% to 60% in several studies. Long-term DFS is strongly influenced by cytogenetic and molecular abnormalities present at diagnosis. Transplant options should be considered for patients with high-risk features while in first remission due to poor outcomes with conventional chemotherapy. Patients with t(8;21), inv(16), or isolated NPM-1, mutations can expect a 60% relapse-free survival following 3 to 4 cycles of HDAC. Molecular mutations are being used to identify patients with normal karyotypes with a high risk (≥ 50%) of relapse for whom multiple cycles of HDAC consolidation therapy will not be sufficient to prevent relapse.
CNS prophylaxis
CNS prophylaxis is not routinely recommended for adult patients with AML. Exceptions for which a screening lumbar puncture should be considered following remission induction therapy include those at high risk for CNS recurrence, ie, patients with a WBC count > 50,000/μL at presentation or those with myelomonocytic or monocytic AML (FAB M4 or M5). Patients receiving HDAC (≥ 7.2 g/m2) for induction or consolidation therapy achieve therapeutic drug levels in the cerebrospinal fluid, obviating the need for intrathecal therapy. Patients given conventional Ara-C doses may be treated with intrathecal methotrexate (12 mg IT) or Ara-C (30 mg IT). Both agents can be combined with hydrocortisone (30 mg IT) for patients with active CNS disease.
Treatment of Refractory or Relapsed AML
Patients who do not respond to initial therapy or who relapse within 6 months of attaining CR, as well as those with antecedent myelodysplasia or therapy-related AML, are considered to have relatively resistant disease.
Efforts to overcome drug resistance have focused on (1) HDAC-containing regimens, (2) new agents, and (3) targeted therapy using leukemia-specific monoclonal antibodies conjugated with radionuclides or toxins.
HDAC High doses of Ara-C (2 to 3 g/m2 for 8 to 12 doses) paired with mitoxantrone, etoposide,



methotrexate, or fludarabine have produced short-lived CRs in 40% to 60% of relapsed patients with AML (see Table 5 for dosage regimens). Response rates were higher in patients who had received standard-dose Ara-C for induction therapy and who had subsequently relapsed than in those in whom induction therapy had failed. The median duration of remission was 4 to 6 months.
Combinations of mitoxantrone and etoposide have been reported to produce a 40% to 50% CR rate in patients who had relapsed or for whom standard-dose Ara-C and anthracycline had failed, again with a median duration of remission of 4 to 6 months. Combinations of intermediate-dose Ara-C (1 g/m2/day for 6 days) with mitoxantrone and etoposide produced CR rates of 79% in relapsed patients and 46% in those who did not respond to induction therapy or had AML evolving from MDS, with a median CR duration of 8 months.
New agents Nucleoside analogs, such as cladribine (2-CdA) and fludarabine, showed activity in pediatric AML. A British trial reported a 61% CR rate for a combination of fludarabine, Ara-C, G-CSF, and idarubicin, with a median CR duration of 7 months. In an Italian study, 30 patients < 65 years of age received fludarabine, Ara-C, and idarubicin combined with gemtuzumab ozogamicin (3 mg/m2 on day 6). The CR rate was 90%, with two cases of resistant disease and one death during induction therapy. Following 1 cycle of consolidation therapy, 19 of 26 patients received allogeneic HCT. With a median follow-up at 16 months, 24 patients (80%) remain alive and in remission. Clofarabine showed a 16% remission rate in a phase I/II trial in patients with relapsed AML as a single agent and a 38% remission rate when combined with Ara-C (1 g/m2).
Temozolomide (Temodar), an oral alkylating agent currently approved for the treatment of astrocytoma, has been shown to have activity against myeloid malignancy, with a 20% clearance of marrow blasts in 18 patients with high-risk myeloid malignancy following a 7-day course of medication. Prolonged marrow aplasia was the dose-limiting toxicity.
Targeted therapy Gemtuzumab ozogamicin has been approved by the FDA for the treatment of relapsed AML in older patients (Table 5), as mentioned previously. Aggregate data from three trials involving 277 patients, with a median age of 61 years, showed a 26% CR rate, with a median remission duration of 6.4 months.
Treatment-related toxicity was low; the only infusional side effects were fever/chills and slow platelet plus granulocyte recovery (≥ 5 weeks). No cardiac or cerebellar toxicities were reported. Liver function abnormalities were reported in 25% to 30% of patients. The median CR duration for responding patients was 9 months.
Transplantation Although none of the previous options currently offers more than a 10% to 15% chance of long-term DFS, they do provide temporary cytoreduction sufficient to permit further high-dose treatment strategies, such as bone marrow transplantation using sibling, unrelated donor, or purged autologous marrow. Allogeneic bone marrow transplantation achieves a 30% to 40% DFS rate at 5 years in patients transplanted during first relapse or second remission. Autologous bone marrow transplantation also has curative potential for patients beyond first CR, with most large series reporting DFS rates of 30% to 35% in selected patients (usually those with good-risk cytogenetics or initial CR duration longer than 1 year).
Reduced-intensity conditioning regimens are being explored as treatment options in older patients and in those with comorbidity that would otherwise preclude full-dose allogeneic transplantation. Preliminary results from several centers have shown 1- and 2-year DFS rates of 50% for patients aged 55 to 70 years receiving reduced-intensity allogeneic transplantation for consolidation of first remission.
New methods of marrow purging and post-transplant immune stimulation also are being explored to decrease relapse-related mortality.
TREATMENT OF APL
APL represents a uniquely homogeneous subset of AML defined by its cytogenetic abnormality, t(15;17), which results in fusion of the retinoic acid receptor (RAR)α gene on chromosome 17 with the promyelocytic leukemia (PML) gene on chromosome 15. This abnormality yields the PML/RARα fusion protein, detectable by PCR techniques, which is useful for both diagnosis and evaluation of minimal residual disease. Most patients (80%) with APL have characteristic hypergranular blasts; laboratory evidence of DIC is present in 70% to 90% of patients at diagnosis or shortly after. Hemorrhagic events contribute 10% to 15% excess mortality during induction chemotherapy for APL compared with other AML subtypes.
Because of the unique biology and specific clinical features of APL, induction and consolidation regimens for APL differ from strategies used for other FAB types.
Involvement of the RARα gene in the pathogenesis of APL suggested the use of retinoids as therapy. A study from Shanghai showed CR rates of 85% with single-agent all-trans-retinoic acid (ATRA, Tretinoin, Vesanoid). ATRA offered the advantages of a shorter neutropenic period (2 weeks) and slightly faster resolution of DIC (4 vs 7 days), as compared with standard chemotherapy with Ara-C and daunorubicin. Normalization of marrow morphology and cytogenetics requires 30 to 60 days of ATRA.
Initial treatment options
Based on data from the Spanish PETHEMA Group trials, a stratification schema of risk of relapse was constructed using WBC and platelet counts at presentation. Patients with a WBC count < 10,000/μL and a platelet count > 40,000/mL have a DFS of 97%; those with a WBC count < 10,000/μL and a platelet count < 40,000/mL have a DFS of 86%; those with a WBC count > 10,000/μL have a DFS of 78%.
The backbone of APL induction therapy includes an anthracycline and ATRA (Table 6). The French


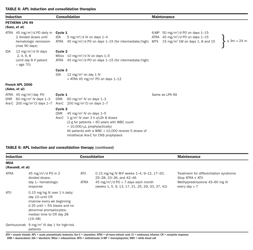
and North American APL trials have also included standard-dose Ara-C as an integral part of induction and consolidation therapies. All three groups report CR rates in excess of 90% in patients with an initial WBC count < 10,000 /μL.
In a comparison of the outcomes from the Spanish LAP 99 trial and the French APL 2000 trial, the CR rates and 3-year survival rates were similar for the low- and intermediate-risk groups, with a lower rate of relapse (4% vs 14%) and fewer days in the hospital (50 vs 72 days) for the group that did not receive Ara-C. In patients with elevated WBC counts ≥ 10,000 μL, Ara-C during induction significantly increased CR rates (95% versus 83%). Survival at 3 years was also higher for the group receiving Ara-C (92% vs 81%), and relapse was lower (9.9% vs 18.8%). In the LAP 99 trial, the use of ATRA along with anthracycline in consolidation significantly decreased the relapse rate among the low- and intermediate-risk groups.
The most recent North American Intergroup trial showed improved relapse-free survival when 2 cycles of arsenic trioxide (Trisenox) were used as the initial component of consolidation. All three groups were monitored for molecular remission at the end of consolidation and at frequent intervals during 2 years of maintenance chemotherapy. Relapse rates are lower than 5% for low-risk patients, and new trials will evaluate the need for maintenance therapy for this group.
Small single-institution series have reported favorable remission and DFS rates in patients induced with arsenic trioxide alone (CR of 86%) or combined with ATRA (95% for low- and intermediate-risk patients). High-risk patients had poorer response rates (CR of 75%) despite the addition of gemtuzumab ozogamicin (9 mg/m2) on day 1 of induction therapy.
APL syndrome Approximately 25% of patients with APL develop “differentiation syndrome” (formerly known as ATRA syndrome). Symptoms of this syndrome are fever, respiratory distress with pulmonary infiltrates or pleural effusions, and cardiovascular collapse. Temporary pseudotumor cerebri is a fairly common (10%) side effect of ATRA. Although these symptoms most often correlate with leukocytosis (WBC count > 10,000/μL), many patients develop symptoms with WBC counts between 5,000/μL and 10,000/μL. The syndrome is seen in patients treated with arsenic trioxide as well as in those treated with ATRA.
Treatment of this syndrome involves prompt use of high-dose steroids, initiation of conventional Ara-C/daunorubicin chemotherapy to control leukocytosis, and temporary discontinuation of ATRA or arsenic trioxide.
Relapse therapy Arsenic trioxide is now the standard reinduction therapy for patients with APL who are refractory to, or have relapsed from, retinoid and anthracycline chemotherapy. As a single agent, arsenic trioxide has produced CR in 34 of 40 patients (85%) with relapsed APL, with 86% of patients achieving molecular remission. Relapsed patients who achieved a molecular remission with arsenic trioxide alone had a median relapse-free survival of 18 months; those who received arsenic trioxide followed by autologous transplantation have had relapse-free survivals in excess of 70% at 2 years. Allogeneic transplantation should be reserved for those who do not achieve a molecular remission.
Although liver toxicity was reported with the use of arsenic trioxide in the original Chinese studies, the most significant toxicities in the US multicenter trial were the “APL syndrome,” ventricular arrhythmia in patients with prolongation of the AT/QTc interval on electrocardiogram, and peripheral neuropathy. It is important to monitor potassium, magnesium, and calcium levels closely, almost daily, during arsenic trioxide therapy; maintaining these levels near the upper range of normal is important in preventing arrhythmia.
Gemtuzumab ozogamicin is also an effective agent for patients with relapsed APL. In a small series, 91% of patients with a molecular relapse of APL achieved a molecular remission following two doses of gemtuzumab ozogamicin (6 mg/m2).
Monitoring response to therapy Reverse-transcriptase PCR for the PML/RARα fusion protein can be used to follow response to therapy. The marker clears slowly, with many patients still testing positive following induction therapy. However, patients with persistence of PML/RARα fusion protein at the end of consolidation therapy are at high risk of relapse, as are those with reemergence of the marker following a period without detectable protein. Salvage chemotherapy should be considered for patients with persistent or recurrent confirmed molecular relapse.
References:
SUGGESTED READING
On All
Bruggemann M, Ruff T, Florhr T, et al: Clinical significance of minimal residual disease quantification in adult patients with standard risk acute lymphoblastic leukemia. Blood 107:1116-1123, 2006.
DeAngelo DJ, Yu D, Johnson JL, et al: Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood 109:5136-5142, 2007.
Gruber F, Mustjoki S, Porkka K: Impact of tyrosine kinase inhibitors on patient outcomes in Philadelphia chromosome-positive acute lymphoblastic leukemia. Br J Haematol 145:581-597, 2009.
Jabbour E, O'Brien S, Kantarjian H, et al: Neurologic complications associated with intrathecal liposomal cytarabine given prophylactically in combination with high-dose methotrexate and cytarabine to patients with acute lymphocytic leukemia. Blood 109:3214-3218, 2007.
Moorman AV, Harrison CJ, Buck GA, et al: Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 109:3189-3197, 2007.
Stock W, La M, Sanford B, et al: What determines outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children's Cancer Group and Cancer and Leukemia Group B studies. Blood 112:1646-1654, 2008.
Storring JM, Minden MD, Kao S, et al: Treatment of adults with BCR-ABL negative acute lymphoblastic leukemia with a modified paediatric regimen. Br J Haematol 146:76-85, 2009.
On AML
Applebaum FR, Gundacker H, Head DR, et al: Age and acute myeloid leukemia. Blood 107:3481-3485, 2006.
Bachner T, Berdal WE, Schoch C, et al: Double induction containing either two or one course of high dose cytarabine plus mitoxantrone and post remission therapy with either autologous stem-cell transplantation or by prolonged maintenance for acute myeloid leukemia. J Clin Oncol 24:2480-2489, 2006.
Candoni A, Martinelli G, Toffolotti E, et al: Gemtuzumab ozogamicin in combination with fludarabine, cytarabine, idarubicin (FLAI-GO) as induction therapy in CD33-positive patients younger than 65 years. Leuk Res 32:1800-1808, 2008.
Lowenberg B, Ossenkoppele GJ, van Putten W, et al: High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 361:1235-1248, 2009.
Schlenk RF, Döhner K, Krauter J, et al: Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358:1909-1918, 2008.
on APL
Ades L, Sanz MA, Chevret S, et al: Treatment of newly diagnosed acute promyelocytic leukemia (APL): A comparison of French-Belgian-Swiss and PETHEMA results. Blood 111:1078-1084, 2008.
Ravandi F, Estey E, Jones D, et al: Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol 27:504-510, 2009.
Sanz MA, Martin G, Gonzalez M, et al: Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: A multicenter study by the PETHEMA group. Blood 103:1237-1243, 2004.
Abbreviations in this chapter
ALSG = Australian Leukemia Study Group; BFM = Berlin-Frankfurt-Munster; CALGB = Cancer and Leukemia Group B; COG = Children Oncology Group ECOG = Eastern Cooperative Oncology Group; GLSG = German Leukemia Study Group MRC = Medical Research Council; SWOG = Southwest Oncology Group














