Chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is a clonal malignancy that results from expansion of the mature lymphocyte compartment. This expansion is a consequence of prolonged cell survival, despite a varied cell. The affected lymphocytes are of B-cell lineage in 95% of cases, and the remaining cases involve T lymphocytes, representing a distinct disorder.
Chronic lymphocytic leukemia (CLL) is a clonal malignancy that results from expansion of the mature lymphocyte compartment. This expansion is a consequence of prolonged cell survival, despite a varied cell. The affected lymphocytes are of B-cell lineage in 95% of cases, and the remaining cases involve T lymphocytes, representing a distinct disorder.
CLL is the most common leukemia in adults in Western countries, accounting for approximately 25% to 30% of all leukemias. The proportion of cases diagnosed with the early stages of the disease (Rai stage 0) has risen from 10% to 50%, probably because of earlier diagnosis (routine automated blood counts).
Epidemiology
The incidence of CLL in the general population is 3.9:100,000 population, with an estimated death rate of 1.1:100,000 population. It was estimated that there were 15,340 patients diagnosed with CLL in 2007.
Gender The male-to-female ratio is 2:1. There is little change with age, as the male-to-female ratio is 2.1:1.0 for patients < 65 years old, compared with 1.9:1.0 for those ≥ 65 years old.
Age The median age at diagnosis is 72 years, and CLL is rarely seen before the age of 35 years. Less than 3% of all cases are younger than 45 years at the time of diagnosis.
Race In the American population, the incidence of CLL is similar in different races. However, the incidence is much lower in Asia (Japan, Korea, and China), Latin America, and Africa than in the United States and Western Europe.
Etiology and risk factors
The etiology of CLL is unclear. However, some factors associated with CLL have been identified.
Genetic factors There is a high familial risk for CLL, with family members of CLL patients having a twofold to sevenfold higher risk of developing the disease. CLL with a familial association tends to occur in younger individuals with subsequent generations, perhaps because of increased screening. Association with certain human lymphocyte antigen (HLA) patterns has not been consistent.
Environmental factors There is no documented association of CLL with exposure to radiation, alkylating agents, or known leukemogenic chemicals. However, exposure to some chemicals used in agriculture may increase the risk of developing CLL.
Viral infections Associations between CLL and several viruses, including human T-cell lymphotrophic viruses I and II (HTLV-I and HTLV-II) and Epstein-Barr virus (EBV), have been suggested. However, no conclusive evidence of a causal relationship exists. Adult T-cell leukemia/lymphoma, a T-cell disorder that can resemble CLL, is caused by HTLV-I.
Monoclonal B lymphocytosis In one study, 3.8% of unselected healthy individuals older than age 65 in the general population had monoclonal (by light chain analysis) CD5+/19+/23+ B cells. These asymptomatic individuals did not have lymphocytosis or clinical evidence of disease and did not fulfill diagnostic criteria for CLL. Whether or not these individuals will eventually develop diagnostic criteria or symptomatic disease is unknown. Nevertheless, this finding indicates that the incidence of a monoclonal lymphoproliferative process is potentially much more common in the aging population than previously appreciated.
Signs and symptoms
In the majority of patients, CLL is asymptomatic at diagnosis and is discovered on a routine blood examination. When symptoms are present, they are nonspecific and include fatigue, weakness, and malaise and, in fact, may not even be attributable to CLL.
Constitutional B symptoms (ie, fever, weight loss, and night sweats) are not common at diagnosis but may signal disease transformation. Patients frequently notice enlarged lymph nodes or abdominal discomfort and early satiety related to splenomegaly.
Patients with CLL have an increased susceptibility to infections, which may be the presenting complaint.
Lymphadenopathy is common at diagnosis. Lymph nodes are usually symmetrical, mobile, and nontender.
Splenomegaly and hepatomegaly The spleen and, less frequently, the liver may be enlarged. Splenomegaly may be massive in advanced cases. Only occasionally is splenomegaly found in the absence of lymphadenopathy, but recognition of such patients may identify a group that may benefit from splenectomy.
Other organs In advanced disease, other organs may be involved, including the GI mucosa, prostate, lungs, pleura, and bones. Rarely is such involvement clinically important unless (Richter’s) transformation has occurred.
Laboratory features
Peripheral blood The most consistent feature of CLL lymphocytosis, with median values of 30–50 × 109/L. The lymphocytes are small and mature-appearing, with little cytoplasm and clumped chromatin. A few larger nucleolated cells, which represent prolymphocytes, usually constitute < 10% of the total lymphocytes. Diagnostic criteria for CLL defined by the National Cancer Institute (NCI) and International Workshop on CLL (IWCLL) are presented in Table 1.



A positive Coombs’ test is seen in as many as 30% of patients at some time during the disease course, although it is uncommon (< 5%) during early stages. Autoimmune phenomena are relatively frequent, with hemolytic anemia (lifetime risk, approximately 10% to 20%) and thrombocytopenia (lifetime risk, approximately 5% to 10%) occurring most commonly. Autoimmune neutropenia and other autoimmune sequelae are infrequent but more common than in the general population.
Bone marrow The bone marrow is usually hypercellular but can be normocellular. The most characteristic feature is the presence of at least 30% mature lymphocytes. The lymphocyte infiltration can be interstitial, nodular, mixed interstitial and nodular, or diffuse. Diffuse lymphocyte infiltration is associated with a poor prognosis.
Other laboratory findings Progressive hypogammaglobulinemia is seen in > 50% of patients with CLL, usually affecting IgA first, followed by IgM and IgG. However, 5% to 10% of patients may have a small monoclonal peak. Paraproteinemia is more common at disease transformation.
Elevated serum levels of B2-microglobulin (β2M) have been associated with a poor prognosis. Elevation of serum lactate dehydrogenase (LDH) levels is found in < 10% of patients at diagnosis and may indicate autoimmune hemolytic anemia or (Richter’s) transformation to large-cell lymphoma (LCL).
Immunophenotyping More than 95% of all cases of CLL have a B-cell phenotype with monoclonality by light chain restriction. In these patients, CD19 and/or CD20 are essentially always coexpressed with CD5, which normally is expressed on T cells and a subset of normal B cells. Other markers, such as CD21 and CD22, may also be expressed. Expression of CD23 helps to differentiate CLL from mantle cell lymphoma, in which cells coexpress CD19 and CD5 but lack CD23. Furthermore, the monoclonal antibody FMC7 (which recognizes an epitope on CD20) rarely reacts with CLL cells but frequently binds the cells of patients with mantle cell lymphoma.
Expression of surface immunoglobulin is usually weak and is lower than in normal B lymphocytes or most other B-cell lymphomas. Expression of CD38 on the surface of CLL cells portends a worse prognosis than that for patients whose cells do not express CD38.
Cytogenetic and molecular findings
Chromosomal abnormalities
A number of recurrent cytogenetic abnormalities have been identified in CLL. Most have not been associated with a specific gene defect or abnormality; however, their detection is prognostically important, and work continues toward identifying the associated gene or genes.



Chromosomal abnormalities occur in 50% to 65% of CLL patients with analyzable metaphases. Because of the low mitotic rate in CLL, traditional karyotypic methods frequently fail. Fluorescent in situ hybridization (FISH) has increased the detection of clonal genetic abnormalities in CLL patients. In a landmark study, Dohner et al evaluated 325 patients with CLL. Using a variety of fluorescent probes, they identified chromosomal aberrations in 82%. Among these findings was the recognition that some subtypes (17p del and 11q del) had markedly shorter time to initiate chemotherapy and shorter overall survival than did other types. In this study, the most frequent change was a deletion in 13q14 (55% of patients). Other typical abnormalities included deletion 11q22–23 (18%), trisomy 12q13 (16%), and deletion 17p13 (7%).
These genetic abnormalities help explain some of the clinical variations seen in CLL. For example, patients with 13q deletions tend to have modest or absent lymphadenopathy, whereas patients with deletion 11q frequently have bulky adenopathy.
Disease progression also is heavily influenced by the underlying genetic abnormality. Time from diagnosis to treatment averaged only 9 months for patients with 17p abnormalities, compared with 92 months for patients with 13q deletions.
Prognostic importance These chromosomal abnormalities were potent predictors of outcome with the following median survivals: deletion 17p, 32 months; deletion 11q, 79 months; trisomy 12, 114 months; and deletion 13q, 133 months.
Molecular abnormalities
No single gene has been implicated in the pathogenesis of CLL. However, several genetic abnormalities have biologic and/or prognostic implications.
The p53 gene is located on the short arm of chromosome 17 and is deleted in the leukemia clone of up to 10% of patients with CLL. Mutations of p53 occur in a similar proportion of CLL cases, usually in association with p53 deletion in the other p53 allele. The 17p deletion involving p53 is considered the most significant negative cytogenetic prognostic factor in CLL. The p53 protein normally responds to DNA damage by inducing cell cycle arrest and facilitating DNA repair. It can also induce apoptosis in cells with damaged DNA and in this way mediates the cytotoxicity of many anticancer agents. Resistance to treatment is a particular characteristic of p53 deletion and has been observed for agents including purine analogs.
Overexpression of bcl-2 Abnormalities of the long arm of chromosome 14 frequently involve region 14q32, the site encoding for the immunoglobulin heavy-chain gene. However, gene translocations such as t(11;14)(q13;q32) and t(14;18)(q32;q21), which juxtapose genes bcl-1 and bcl-2 to the heavy-chain immunoglobulin gene, are relatively uncommon and should prompt consideration of alternative diagnoses (mantle cell or follicular lymphoma). Nevertheless, increased expression of bcl-2 mRNA and protein is common in CLL. Since overexpression of bcl-2 inhibits apoptosis, it is possible that this gene participates in the pathogenesis of CLL.
Staging and prognosis
Staging systems Two staging systems of CLL are commonly used: one proposed and later modified by Rai and the other proposed by Binet (Table 2). Both systems include three categories of low, intermediate, and high risk, with median survival durations of approximately 10, 6, and 2 years, respectively.


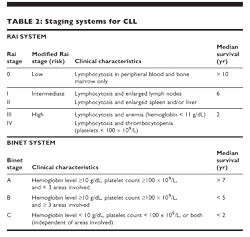
Other prognostic factors Stage of disease has been considered the main prognostic indicator for CLL. However, other factors have prognostic implications, such as chromosomal aberrations, serum level of β2M, pattern of bone marrow infiltration, the lymphocyte doubling time, and serum levels of soluble CD23.
Somatic hypermutation (SHM) normally occurs in germinal centers of lymphoid tissues and is a process that introduces mutations in the variable region of immunoglobulin genes. It is responsible for affinity maturation, the process of producing B cells with high-affinity antibody. CLL can be divided into two distinct prognostic groups based on the extent to which the expressed immunoglobulin heavy chain variable (IgVH) gene has undergone SHM. CLL cases with an IgVH gene with < 98% homology to germline are considered mutated and those with an IgVH gene with ≥ 98% homology to germline are considered unmutated.
CLL cases with an unmutated IgVH gene generally have rapid progression and an unfavorable prognosis, whereas CLL cases with a mutated IgVH gene generally have slowly progressive disease and a favorable prognosis. Patients with unmutated IgVH genes have a median survival of 8 years, compared with 24 years for those with mutated IgVH genes. The biologic differences between these groups are also reflected in the association of unmutated IgVH genes with other poor prognostic features, such as expression of CD38, and unfavorable cytogenetic abnormalities. These results need to be interpreted carefully in light of the fact that the study population was not representative of the epidemiology of CLL, where the median survival for all patients is approximately 5 years.
The immunoglobulins expressed by CLL cells can be polyreactive autoantibodies. It follows that the cell that gives rise to the CLL B cell may experience persistent antigen stimulation, which contributes to malignant transformation. The role of antigen selection is implicated by the overrepresentation of certain IgVH genes in CLL. In addition, there is also a biased distribution of these genes between the mutated and unmutated subgroups. The VH3 and VH4 families of genes are frequently found in the subgroup with mutated IgVH genes, whereas the VH1 family, particularly VH1-69, is more common in the subgroup with unmutated IgVH genes. Specific genes, such as VH3-21, have also been associated with a poor prognosis, independent of IgVH mutation status.
ZAP-70 encodes a protein tyrosine kinase normally expressed by T cells and involved in intracellular signaling initiated by T-cell receptor ligation. Expression of ZAP-70 by CLL B cells (> 20% of CLL are cells ZAP-70 positive) is associated with unmutated IgVH genes, shorter time to initial treatment, and inferior survival compared with ZAP-70 negative (< 20% cases). Detection of ZAP-70 protein by flow cytometry or immunohistochemistry may provide a useful prognostic marker for patients with CLL.
Treatment
TREATMENT RECOMMENDATIONS
Traditionally, the initial therapy for CLL has been chlorambucil (Leukeran) with or without prednisone. However, accumulating data suggest that fludarabine has significantly greater activity than these agents. This agent produces higher overall and complete remission (CR) rates and provides a longer remission duration than chlorambucil. Newer data suggest that combination chemotherapy, particularly a nucleoside analog combined with an alkylating agent, produces higher response rates, and the addition of monoclonal antibodies to such regimens appears to increase the CR rates. However, to date, a survival advantage has not been demonstrated for administering these regimens as initial therapy.
The development of nonmyeloablative transplants has provided the possibility of allogeneic transplantation for CLL, where the median age of patients is near 70, but this new technique should only be performed in the context of a clinical trial. An effort should be made to enroll patients in clinical trials that offer them the possibility of receiving some of the new alternatives, which may eventually achieve the goal of curing CLL.
EARLY-STAGE DISEASE



Since patients with early-stage CLL have a good long-term prognosis, and early therapy has not changed the outcome of the disease, patients in the early stages should not be treated unless specific indications exist (Table 3). Randomized trials comparing chlorambucil versus no therapy have documented no advantage for patients with early-stage CLL who received immediate therapy with chlorambucil. New alternatives for treatment of high-risk early-stage CLL (ZAP-70–positive, unmutated IgH status) are being investigated (eg, monoclonal antibodies).
CONVENTIONAL CHEMOTHERAPY
Single-agent chemotherapy



Chlorambucil Historically, the initial agent used for CLL has been chlorambucil, given as either 0.1 mg/kg daily or 20 to 40 mg/m2 every 2 to 4 weeks. Therapy is continued until the signs or symptoms requiring therapy are controlled.
Chlorambucil is frequently combined with oral prednisone (30 to 100 mg/m2/d), although there is no clear evidence that the combination improves responses or overall survival over chlorambucil alone. Prednisone is of value, however, in the management of autoimmune cytopenias.
Cyclophosphamide is an alternative to chlorambucil. The usual dose is 0.5 to 1 g/m2 every 3 to 4 weeks alone or together with vin-cristine and steroids (eg, COP [cyclophosphamide, vincristine (Oncovin), and prednisone] regimen).
Combination chemotherapy
COP and CHOP Various drug combinations have been used in CLL, mostly in patients with advanced-stage disease. The most frequently employed combinations have been COP and these three drugs plus doxorubicin (CHOP). The dose of doxorubicin used is usually low (25 mg/m2). A higher dose of doxorubicin (50 mg/m2) has been employed in some regimens, such as CAP (cyclophosphamide, doxorubicin [Adriamycin], and prednisone).
Response rates have been 40% to 85% with these combinations. In randomized studies, COP was no better than chlorambucil plus prednisone. Although CHOP initially achieved better survival than COP (in patients with Binet stage C disease) or chlorambucil plus prednisone, longer follow-up has not confirmed this survival advantage.
Nucleoside analogs



Fludarabine, now frequently the drug of choice for treating CLL, has been demonstrated to be more effective than chlorambucil for the treatment of CLL. When given to previously treated patients at a dose of 25–30 mg/m2/d for 5 days every 3 to 4 weeks, this nucleoside analog produced responses in 20% to 50% of patients, with 5% to 15% of patients achieving CR and an additional 5% to 20% achieving a “nodular partial response (PR),” ie, a CR but with the presence of lymphoid nodules in the bone marrow (Table 4). In previously untreated patients, the response rate was 63% to 80%, with 8% to 35% of patients achieving a CR.
The addition of prednisone or chlorambucil to fludarabine therapy did not improve the response rate and is associated with an increased incidence of opportunistic infections and other toxicities. A large randomized study comparing fludarabine, CAP, and CHOP demonstrated an increased response rate with fludarabine but no difference in survival (Table 5). Randomized trials of fludarabine versus chlorambucil in previously untreated patients showed improvements in response rate (overall and CR), duration of response, and disease progression-free survival with fludarabine but no survival advantage.



Other nucleoside analogs Cladribine (2-chlorodeoxyadenosine, 2-CdA) is also active in CLL when given at doses of 0.1 mg/kg/d (or 4 mg/m2/d) for 7 days. At therapeutic doses, this agent appears to be associated with more myelosuppression, particularly thrombocytopenia, than fludarabine. This finding limits its utility in treating CLL, but a direct comparison with fludarabine has not been reported.
The third purine analog active against CLL is pentostatin (Nipent). Previously, toxicity limited its use as an antineoplastic agent. More recently, the recognition that safe use of this drug requires concomitant hydration and close attention to renal function (it is both toxic to and cleared by the kidneys) has renewed interest in clinical evaluation with this agent. The group at Memorial Sloan-Kettering Cancer Center has studied pentostatin combined with cyclophosphamide and demonstrated responses in > 70% of previously treated patients (including those whose disease is refractory to fludarabine) with acceptable toxicity.
NEW APPROACHES



Combination chemotherapy The combination of fludarabine, cyclophosphamide, and rituximab (Rituxan) has been studied by the group at M. D. Anderson. In 224 previously untreated patients, the overall response rate was 95%, with 70% achieving a CR and two-thirds of patients achieving a flow CR.
Using a sequential approach with these two agents, the group at Memorial Sloan-Kettering Cancer Center has shown that consolidation therapy with high-dose cyclophosphamide with and without rituximab markedly improves the frequency of a CR over fludarabine alone.
Data suggest that the addition of a monoclonal antibody to fludarabine-based therapy may markedly improve CR rates in this disease. A study conducted by the Cancer and Leukemia Group B (CALGB) randomized patients with previously untreated CLL to receive fludarabine or a combination of fludarabine and the anti-CD20 antibody rituximab at standard doses. Patients in both arms subsequently received a consolidation course of rituximab for 4 weeks.
Overall response rates were high in both arms, but there was a significantly higher CR rate (33%) in the concurrent arm than in the arm with fludarabine alone (15%); however, both groups experienced similar response durations and overall survival. The CR rate increased in both groups after consolidation therapy with rituximab. This study illustrated the fact that adding rituximab to fludarabine increases the frequency of a CR both when administered concomitantly and sequentially.
Elter et al reported on the use of fludarabine in combination with alemtuzumab (Campath), a humanized monoclonal antibody targeting the panlymphocyte antigen CD52, in 36 patients with relapsed or refractory disease. The overall frequency of response was 83%, with 11 complete responses.
Monoclonal antibody–targeted therapy Monoclonal antibodies have been used in patients with CLL to exploit antibody-mediated cytotoxicity. Alemtuzumab has been approved by the US Food and Drug Administration (FDA) for the treatment of refractory CLL. In a pivotal trial in patients with fludarabine-refractory disease, alemtuzumab resulted in an overall response rate of 33%.



Rituximab also has been investigated and is active both as a single agent and in combination with chemotherapy. Because of rituximab’s efficacy without significant toxicity, it is assuming a greater role in the treatment of patients with CLL.
The combination of alemtuzumab and rituximab has been evaluated in patients with relapsed or refractory lymphoid malignancies. Alemtuzumab was given twice weekly at 30 mg IV for 4 weeks. Rituximab (375 mg/m2) was given concurrently weekly for the 4 weeks. Responses were reported for 48 patients, 32 with CLL, 9 with CLL/PLL (prolymphocytic leukemia), 1 with PLL, 4 with mantle cell lymphoma, and 2 with Richter’s transformation. The overall response rate was 52%; CR was noted in 8%, nodular PR in 4%, and PR in 40% of treated patients. Toxicities included infusion-related reactions. Infections occurred in 52% of patients with cytomegalovirus reactivation occurring in 27% of patients. Overall, this was a well-tolerated combination for relapsed patients with CLL.
Achieving minimal residual disease-free status The current formal criteria to assess response are based on physical examination, blood counts, and microscopic evaluation of bone marrow (Table 4). Minimal residual disease (MRD) can be evaluated by more sensitive methods such as polymerase chain reaction (PCR) for the IgVH gene or 4-color flow cytometry. In a study of 91 previously treated patients who received alemtuzumab (30 mg thrice weekly for up to 16 weeks), 36% achieved CR, and the overall response rate was 54%. Of the 91 patients, 18 achieved MRD-free status by flow-cytometry evaluation of their bone marrow. Survival was significantly longer for patients who achieved MRD-free status. Overall survival for the 18 patients with MRD-free remission was 84% at 5 years.
For patients who have residual disease after purine analog–based therapy, the marrow has been the major site of involvement. Elimination of residual disease, particularly in the marrow, may improve treatment outcome. Because of the significant activity of alemtuzumab at clearing blood and marrow of disease, its ability to eliminate residual disease after chemotherapy is under investigation.
Stem-cell transplantation
Both allogeneic and autologous stem-cell transplantations (SCTs) have been tested in patients with CLL.
Allogeneic SCT is a viable option for younger patients with CLL, particularly if they have not responded to alkylating agents and/or nucleoside analogs and are in an advanced stage of disease. The series reported to date, including a majority of patients with advanced refractory disease, has documented a CR rate in excess of 70%. The response is sustained in most patients, although reported follow-up is typically short. SCT using nonablative conditioning regimens has produced encouraging results and should be considered in the setting of a clinical trial, particularly for patients > 60 years.
Autologous SCT Since the median age of patients with CLL is usually higher than the age considered acceptable for allogeneic SCT, autologous transplants using purged marrow have also been investigated. In general, results have been disappointing, with the best results seen in patients with responsive disease and low tumor burdens-a group that might have fared well without a transplant.
Splenectomy
Splenectomy may be beneficial for patients in whom hypersplenism is the cause of cytopenias (particularly in patients without significant lymphadenopathy) or for palliation when splenomegaly is symptomatic and refractory to chemotherapy. Cytopenias frequently respond to splenectomy. Perioperative mortality varies widely and is largely related to the experience of the surgeon in performing splenectomy in these patients. In experienced hands, splenectomy can be performed with minimal mortality, even in patients with end-stage disease.
Complications
Infections Patients with CLL are prone to multiple infections, and infectious complications are a leading cause of death in patients with CLL. Hypogammaglobulinemia plays a central role in the predisposition of patients to this problem, and prophylactic IV administration of immunoglobulin preparations may reduce the incidence of infections. Cytotoxic therapy further weakens the immune system and may increase the risk of opportunistic infections.
Autoimmune cytopenias frequently complicate CLL and may be precipitated or aggravated by therapy (eg, fludarabine) for CLL. Autoimmune hemolytic anemia can be treated successfully with prednisone in the majority of patients. Combinations of cyclophosphamide with rituximab are beneficial in cases refractory to prednisone, splenectomy, or cyclosporine. Similar approaches may be useful for autoimmune thrombocytopenia. In a study from M. D. Anderson Cancer Center, 31 patients with CLL and autoimmune anemia or thrombocytopenia received cyclosporine (300 mg/day). Sixty-three percent of patients responded, with a median duration of response of 10 months. No grade 3/4 toxicity was seen.
Pure red cell aplasia and the less commonly amegakaryocytic thrombocytopenia are infrequent complications of CLL, which are mediated by immune mechanisms. Therapy with cyclosporine (3 to 6 mg/kg/d) is frequently effective.
TRANSFORMATION
LCL CLL transforms into LCL in 3% to 10% of patients. This phenomenon, known as Richter’s transformation, has an aggressive presentation with fever and other B symptoms and progressive lymphadenopathy. Extranodal involvement occurs in approximately 40% of patients. Paraproteinemia, hypercalcemia, and a sharp rise in serum LDH levels can be frequently seen.
The prognosis of patients whose disease progresses to LCL is variable and depends in part on the degree of prior treatment used for the underlying CLL. In treatment-naive patients with LCL, standard therapy with CHOP and rituximab may offer long-term control of the transformed component. In patients who have had significant prior therapy, the disease is often refractory, and combination chemotherapy including SCT is frequently ineffective.
PLL More rarely, CLL can transform into PLL, characterized by a > 55% increase in prolymphocytes. The transformation is frequently accompanied by progression of splenomegaly, cytopenias, and refractoriness to therapy.
Other diseases Anecdotal cases of CLL evolving into acute lymphocytic leukemia, myeloma, low-grade lymphoma, and Hodgkin lymphoma have been reported. The rarity of these reports makes a true causal connection less likely.
HAIRY-CELL LEUKEMIA
Hairy-cell leukemia (HCL) is an infrequent B-cell malignancy usually associated with pancytopenia and splenomegaly. About 600 cases are reported yearly in the United States. Despite its relative rarity, there are a disproportionate number of highly effective therapies available.
Epidemiology and etiology
The male-to-female ratio of HCL is 4:1. The median age at presentation is 50 years. The etiology is unknown.
Differential diagnosis
HCL can be confused with malignant lymphomas, splenic lymphoma with villous lymphocytes (SLVLs), CLL, other non-Hodgkin’s lymphomas in leukemic phase, and occasionally even myelodysplastic syndromes.
Treatment
The indications for treatment of HCL are an absolute neutrophil count (ANC) < 1,000/µL, platelet count < 100 × 103/µL, or hemoglobin level < 10 g/dL; leukemic phase of HCL; symptomatic splenomegaly; recurrent infections; or autoimmune complications.
Response criteria The criteria for a CR are normalization of the complete blood cell (CBC) count, with ANC > 1,500/µL, platelet count > 100,000/µL, and hemoglobin level > 12 g/dL; regression of organomegaly to normal; and bone marrow and peripheral blood free of hairy cells. PRs require reduction of the hairy cells in the bone marrow to < 50%, < 5% hairy cells in peripheral blood, > 50% reduction in organomegaly, and normalization of the CBC count.
Splenectomy is reserved for patients with splenic rupture, infarcts, a massively enlarged spleen, severe hypersplenism, or failure to respond to systemic chemotherapy.
Interferon-α (IFN-α), at a dose of 3 mU/d administered by IM or SC injection for 6 months followed by 3 mU/d three times weekly for 12 or 24 months, induces a CR in 8% to 10% of patients and a PR in 74%. The median time to response was 6 months in patients achieving a PR and 14 months in those achieving a CR. Patients frequently relapse between 12 and 24 months after discontinuation of therapy. The superiority of purine analog therapy (discussed later) has essentially led most clinicians to abandon the use of IFN for this disease.
Purine analogs The recommended dose of pentostatin is 4 mg/m2 by IV bolus every other week until a CR is obtained. Usually, patients require a median of 8 courses (range, 4–15). The CR rate varies between 59% and 89% in different studies, and the PR rate varies between 4% and 37%. Responses can last for many years, and patients who relapse often respond to retreatment with pentostatin. In an update of a large randomized trial comparing pentostatin and IFN-α, Flinn et al reported that in 241 patients with HCL treated with pentostatin, the 10-year overall survival rate was 81%, with only two deaths (1%) attributable to HCL.
Cladribine shows activity in treating HCL similar to that of pentostatin. Due to this finding and the fact that cladribine is given as 1 cycle of a 7-day continuous infusion or a 5-day bolus, this agent usually is the preferred treatment of this disorder. Piro et al treated 144 HCL patients with cladribine,
0.1 mg/kg/d by continuous IV infusion for 7 days. A total response rate of 97% was obtained, with 85% CRs and 12% PRs. Response was independent of previous treatment with IFN or splenectomy, and three patients whose disease was refractory to pentostatin responded to cladribine. Recovery of CBC counts occurred on average by day 61 (range, 11–268 days).
The largest series reporting long-term follow-up results on patients with HCL treated with cladribine was from the Scripps group. A total of 349 patients, with a median duration of response follow-up of 59 months, were evaluated. Twenty-six percent had relapsed at a median of 29 months, but most of them were patients who had achieved only a PR. The time-to-treatment failure rate at 48 months was only 16% in complete responders.
Else et al reported on a large series of 219 patients with HCL, comparing their experience with pentostatin and cladribine. Treatment results were similar in terms of frequency of CR, relapse, and overall survival. Though the results are excellent, with more than 95% of patients alive at 10 years, the disease-free survival curves do not plateau, indicating these drugs are not curative in HCL.
Immunotoxin The NCI evaluated a recombinant immunotoxin containing an anti-CD22 variable domain fused to a truncated Pseudomonas exotoxin; it was given by IV infusion every other day for a total of three doses. Sixteen patients whose disease was resistant to cladribine were treated; 2 achieved a PR and 11 achieved a CR. Of the 11 patients, 3 who had a CR relapsed and were retreated; all of these patients achieved a second CR.
SUGGESTED READING
Byrd JC, Peterson BL, Morrison VA, et al: Randomized phase II study of fludarabine with concurrent vs sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: Results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 101:6–14, 2003.
Calin GA, Ferracin M, Cimmino A, et al: A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353:1793–1801, 2005.
Catovsky D, Richards S, Matutes E, et al: Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): A randomised controlled trial. Lancet 370:230–239, 2007.
Crespo M, Bosch F, Villamor N, et al: ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med 348:1764–1775, 2003.
Damle RN, Wasil T, Fais F, et al: Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94:1840–1847, 1999.
Eichhorst BF, Busch R, Hopfinger G, et al: Fludarabine plus cyclophosphamide versus fludarabine alone in first-line therapy of younger patients with chronic lymphocytic leukemia. Blood: 107:885–891, 2006.
Else M, Ruchlemer R, Osuji N, et al: Long remissions in hairy cell leukemia with purine analogs. Cancer 104:2442–2448, 2005.
Flinn IW, Neuberg DS, Grever MR, et al: Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J Clin Oncol 25:793–798, 2007.
Hamblin TJ, Davis Z, Gardiner A, et al: Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94:1848–1854, 1999.
Hillmen P, Skotnicki AB, Robak T, et al: Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 25:5616–5623, 2007.
Kay NE, Geyer SM, Call TG, et al: Combination chemoimmunotherapy with pentostatin, cyclophosphamide, and rituximab shows significant clinical activity with low accompanying toxicity in previously untreated B chronic lymphocytic leukemia. Blood 109:405–411, 2007.
Keating MJ, O’Brien S, Albitar M, et al: Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as intital therapy for chronic lymphocytic leukemia. J Clin Oncol 23:4079–4088, 2005.
Lamanna N, Kalaycio M, Maslak P, et al: Pentostatin, cyclophosphamide, and rituximab is an active, well-tolerated regimen for patients with previously treated chronic lymphocytic leukemia. J Clin Oncol 24:1575–1581, 2006.
Lundin J, Kimby E, Bjorkholm M, et al: Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 100:768–773, 2002.
Moreton P, Kennedy B, Lucas G, et al: Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol 23:2971–2979, 2005.
Rai KR, Peterson BL, Appelbaum FR, et al: Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med 343:1750–1757, 2000.
Schetelig J, Thiede C, Bornhauser M, et al: Evidence of a graft-versus-leukemia effect in chronic lymphocytic leukemia after reduced-intensity conditioning and allogeneic stem-cell transplantation: The Cooperative German Transplant Study Group. J Clin Oncol 21:2747–2753, 2003.
Wierda W, O’Brien S, Wen S, et al: Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J Clin Oncol 23:4070–4078, 2005.









How Supportive Care Methods Can Improve Oncology Outcomes
Experts discussed supportive care and why it should be integrated into standard oncology care.



