Ocular melanoma is a rare but potentially devastating malignancy arising from the melanocytes of the uveal tract, conjunctiva, or orbit; it represents less than 5% of all melanoma cases in the United States. The management of ocular melanoma varies depending on its anatomic origin, since uveal and conjunctival melanoma have distinct biologies and thus different treatment strategies. Uveal melanoma is the most common type of ocular melanoma and is characterized by activation of the mitogen-activated protein kinase (MAPK) pathway (among other signaling pathways) via mutations in GNAQ or GNA11. Despite primary radiation or surgical therapy, up to 50% of patients will eventually develop metastatic disease, for which there is no standard therapy and no treatment that has been shown to improve overall survival. The biology of conjunctival melanoma is less well characterized but has been associated with BRAF and NRAS mutations, and results in metastatic disease in 20% to 30% of cases. Clinical trials are currently ongoing to further evaluate and optimize the role of targeted therapies, as well as immunotherapies, as both adjuvant and metastatic treatment in uveal and conjunctival melanoma.
Introduction
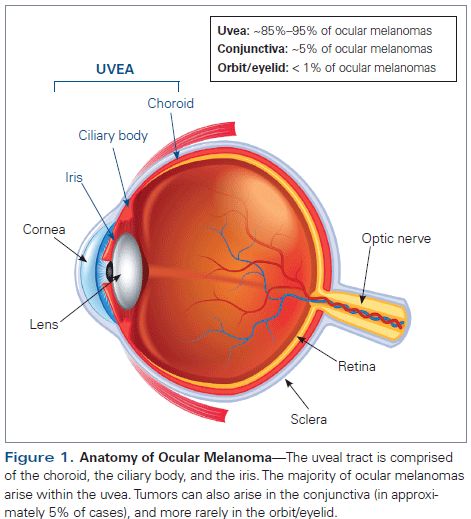
Although the majority of melanomas arise from melanocytes within the skin, they can less commonly arise at other sites. Ocular melanoma is a rare subtype of melanoma that can arise from melanocytes anywhere within the eye, including the uveal tract, conjunctiva, and orbit. Uveal melanoma is the most common type of ocular melanoma; the majority of cases (approximately 85%) occur in the choroid, with the remaining cases arising in the iris or ciliary body (Figure 1).[1] In this review, we discuss current management strategies, as well as future directions, for the management of uveal and conjunctival melanoma.
Uveal Melanoma
Epidemiology and risk factors
Uveal melanoma is the most common primary intraocular malignancy in adults, accounting for about 85% to 95% of ocular melanoma cases. However, uveal melanomas represent only about 3% to 5% of all melanoma cases in the United States.[1,2] While the rate of cutaneous melanoma continues to rise, the incidence of uveal melanoma has remained unchanged. A Surveillance, Epidemiology, and End Results (SEER) database analysis of uveal melanoma cases in the United States from 1973 to 2008 reported a stable incidence of 5.1 per million, with a stable 5-year survival rate of 81.6%.[2] The incidence of uveal melanoma in Europe was found to vary by country, with higher incidences associated with higher geographic latitude (2 per million in Spain and southern Italy; 4–5 per million in France, the Netherlands, Switzerland, and Germany; and > 6 per million in the United Kingdom).[3]
In the United States, uveal melanoma is more common among men than women, and more common among Caucasians than non-Caucasians: the annual age-adjusted incidence per million was 0.31 in African-Americans, 0.39 in Asians, 1.67 in Hispanics, and 6.02 in non-Hispanic whites.[1,4]
The relationship between ultraviolet (UV) light exposure and uveal melanoma risk has not been clearly established. A meta-analysis found that welding was a significant risk factor for uveal melanoma, but chronic UV exposure, including sunlight exposure and geographic latitude, was not significantly related.[5] UV-B radiation exposure and lack of use of sunglasses have been associated with uveal melanoma risk.[6] Other known risk factors for uveal melanoma include: fair skin, light eye color, ability to tan, ocular melanocytosis (a congenital pigmentary abnormality that results in excess melanocytes in the periocular skin or parts of the eye), dysplastic nevus syndrome, and the presence of BAP1 mutations.[7-9]
Molecular biology
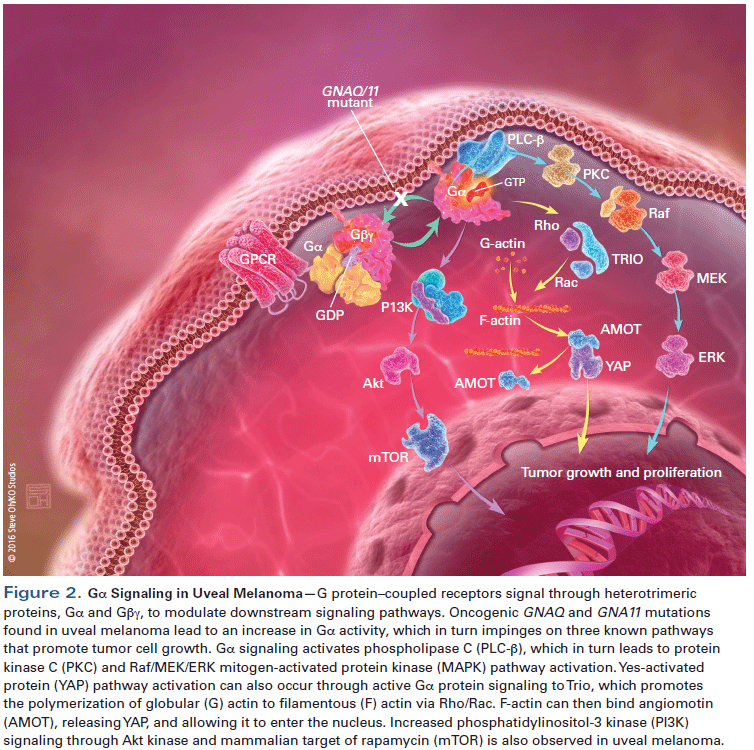
The molecular pathogenesis of uveal melanoma is distinct from that of cutaneous melanoma. While the majority of cutaneous melanomas are associated with activation of the mitogen-activated protein kinase (MAPK) pathway, via activating mutations in BRAF (~50% of cases) or RAS (10% to 25% of cases), or loss-of-function mutations in NF1 (14% of cases),[10] uveal melanoma is characterized by point mutations in the G protein α-subunits GNAQ and GNA11.[11,12] Mutations in GNAQ and GNA11 are found in about 80% of patients and are mutually exclusive.[10-12] These G protein α-subunits are normally inactivated by intrinsic GTPase activity; however, mutations in GNAQ and GNA11 disable their GTPase activity and result in constitutive activity, leading to activation of pathways that include the MAPK and phosphatidylinositol 3-kinase (PI3K)/Akt pathways.[13] GNAQ was recently shown to stimulate the transcriptional coactivator Yes-associated protein (YAP) through the Trio–Rho/Rac signaling circuit, which promotes the polymerization of globular actin to filamentous actin, independent of the canonical Hippo pathway. Filamentous actin then binds to the cytoskeletal protein angiomotin, which displaces YAP and enables it to translocate to the nucleus and initiate transcription of genes involved in proliferation, anti-apoptosis, and cell survival (Figure 2).[14,15]
International efforts such as The Cancer Genome Atlas Project (TCGA) will soon provide the first comprehensive characterization of ocular melanoma by means of combined whole-exome sequencing, RNA sequencing, methylation profiling, and proteomic profiling. Results from these studies will provide insights into the complex genetic and epigenetic changes that occur in ocular melanomas.
Detection and diagnosis of uveal melanoma
Uveal melanoma patients typically present with visual symptoms, including blurred vision, photopsia, floaters, visual field reduction, visible tumor, and pain. Approximately 30% of patients are asymptomatic at diagnosis, and these patients are less likely to undergo enucleation.[16]
Unlike other malignancies, diagnosis of uveal melanoma may often be made by examination alone and is routinely made by a clinical fundoscopic exam without the need for histopathology confirmation. Specialized ocular imaging, including ultrasound and fluorescein angiography, are often performed for further characterization of the disease and prognosis. Biopsy for molecular analysis of the tumor is becoming more routine; however, the results seldom impact the initial treatment plan.
The differential diagnosis includes pigmented nevi and metastases to the uveal tract, as well as other less common entities. Pigmented nevi can be difficult to distinguish from early melanoma by examination. A retrospective study of 2,514 patients with pigmented nevi found that 7% of these cases progressed to melanoma.[17] Several factors, including lesion thickness, subretinal fluid accumulation, vision disturbances, presence of orange pigment on fundoscopic exam, absence of halo, hollow appearance on ultrasound, and a lesion location of less than 3 mm from the optic nerve, were associated with increased transformation risk. Although no standard guideline recommendation exists regarding treatment vs observation of pigmented nevi, most clinicians favor surveillance at least every 6 months to monitor for signs of transformation-although more frequent intervals can be considered for higher-risk lesions.
Prognosis and risk stratification
Several factors have been associated with poor outcome. These include advanced patient age at time of diagnosis; large tumor size; extrascleral extension of the tumor; involvement of the ciliary body; certain pathologic features (eg, epithelioid cytology, extracellular matrix patterns, immune cell infiltration); and incomplete local control after primary tumor treatment.[17-20]
The 2010 American Joint Committee on Cancer (AJCC) tumor node metastasis (TNM) staging system has also been validated for uveal melanoma in a single-institution study.[21] However, classification of tumors based on a 15-gene expression panel showed improved risk stratification compared with TNM staging.[22,23] The use of this gene expression profile relies on a biopsy sample and classifies patients into low metastatic risk “Class 1” or high metastatic risk “Class 2.” Patients with high-risk clinical features and/or Class 2 tumor designation can be considered for adjuvant clinical trials and more intensive surveillance following definitive treatment. Although there is no clear survival benefit from earlier detection of metastatic disease,[24] patients could benefit from clinical trial eligibility and palliative therapy with earlier detection.
Cytogenetic alterations have been found to be prognostic in uveal melanoma. Monosomy of chromosome 3 and gains of 8 are associated with a poor prognosis and metastatic disease. Sequencing of uveal melanoma samples with monosomy 3 led to the discovery of inactivating somatic mutations in BAP1 on chromosome 3p21.1.[9] BAP1 mutations are reported in about 50% of uveal melanoma cases and are associated with metastasis.[25] Mutations associated with a more favorable prognosis include SF3B1 and EIF1AX mutations, reported in 18% and 24% of cases, respectively.[26,27]
Primary treatment of uveal melanoma
The goal of treatment of localized uveal melanoma is to preserve vision and prevent metastasis; however, to date, the selection of primary therapy modality has not been demonstrated to affect the risk of distant disease spread or overall survival (OS). Treatment of the primary tumor is guided by tumor size, lesion location, general health of the patient, and patient preference. Although modalities such as charged-particle radiotherapy, proton beam therapy, and surgical excision have been shown to be effective,[28-31] the majority of cases in the United States are treated with plaque brachytherapy.
Brachytherapy. Brachytherapy involves suturing a radioactive plaque to the episclera to deliver a fixed dose directly to the tumor. The two most common radioisotopes used in the plaques are iodine-125 (125I) and ruthenium-106 (106Ru). 125I is generally preferred in the United States, whereas 106Ru is the most widely prescribed plaque in the United Kingdom. 125I plaques emit gamma radiation, which has a deeper penetration than the beta-emitting 106Ru but increased toxicity to surrounding healthy tissue.[32] The lower penetration depth of 106Ru makes it unsuitable for thick tumors, and its use is generally restricted to those that are less than 6 mm in apical thickness.[28,33] Support for the use of 125I brachytherapy comes from the Collaborative Ocular Melanoma Study (COMS) reports of medium-sized tumors treated with either enucleation or 125I brachytherapy.[29]
Regular ophthalmologic examinations should be performed following plaque brachytherapy to assess for radiation-induced damage, including radiation retinopathy, papillopathy, exudative retinal detachment, and cataract, which can develop 2 to 5 years following initial treatment.
Charged-particle radiation therapy. Charged-particle irradiation (with protons, carbon ions, or helium ions) is used to treat medium to large tumors that may not be good candidates for brachytherapy. Long-term outcomes from a randomized trial comparing charged-particle proton therapy to 125I brachytherapy in choroidal or ciliary body melanoma revealed significantly improved local control, eye preservation, and disease-free progression with charged-particle therapy.[34]
Photocoagulation, transpupillary thermal therapy, and photodynamic therapy. Photocoagulation uses a focused laser beam to destroy vessels that supply blood to tumor cells. A high rate of complications is associated with this technique, and a more recent variation using infrared lasers-called transpupillary thermal therapy (TTT)-has been developed. The infrared radiation is only capable of penetrating the surface tumor layer and is therefore suitable for treating small tumors or marginal recurrences following proton therapy. Several studies have explored the potential role of TTT in combination with brachytherapy (known as “sandwich therapy”) and proton beam therapy, with mixed results.[33,35] TTT has also been used as an adjuvant therapy, although no improved local control was observed in 125I-treated juxtapapillary uveal melanoma.[36] Photodynamic therapy involves injection of a light-sensitive compound, such as benzoporphyrin or verteporfin, followed by exposure to a specific wavelength of light in order to release damaging free radicals that can kill nearby vascular endothelial cells. The long-term local control and disease-free survival benefit of this technique are still under investigation.
Surgery. Historically, enucleation was the treatment of choice. There has been a shift towards vision-sparing treatments following the results of COMS, in which enucleation did not seem to provide any survival benefit over 125I brachytherapy in medium-sized choroidal tumors. The 10-year mortality rate was 18% following enucleation compared with 17% following 125I brachytherapy.[29] However, enucleation is still the treatment of choice in select cases, including when there is little probability of retaining vision, the tumor is too large to treat by other methods, a patient presents with extensive bleeding, there is retinal detachment, or the patient has indicated a preference for enucleation. Pre-enucleation radiotherapy did not seem to improve survival in patients with large choroidal melanomas.[37] The COMS quality-of-life report found that patients who underwent enucleation had less anxiety during follow-up visits than patients treated with brachytherapy,[38] which could be a deciding factor when discussing treatment options with patients.
Local resection of the tumor can be performed either by a transretinal (endoresection) or transscleral (exoresection) approach. These surgeries, like enucleation, can have complications, including vitreous hemorrhage and retinal detachment. In addition, there is some concern of local recurrence and iatrogenic tumor spread resulting from the surgeries, although these may be minimized by adjuvant radiotherapy. The use of adjuvant radiotherapy to reduce local recurrence, a risk factor for metastasis, has been more extensively studied in conjunction with transscleral resection.[39,40] The adjunctive role of radiotherapy in transretinal resection remains controversial.[32]
Adjuvant therapy
Local treatment of a primary uveal tumor is effective in preventing local recurrence in over 95% of cases, yet nearly 50% of patients will develop metastatic disease in a median time of 5 years. This pattern of disease progression suggests that subclinical micrometastases are present at the time of diagnosis. Circulating uveal tumor cells have been detected at diagnosis in patients with no detectable metastases.[41] Uveal melanoma metastasizes hematogenously and very rarely spreads to regional lymph nodes unless there is extraocular tumor extension to the episclera. This may be explained by the absence of lymphatic vessels in the eye-apart from the extraocular conjunctiva and limbus-and by the tumor microenvironment.[42,43]
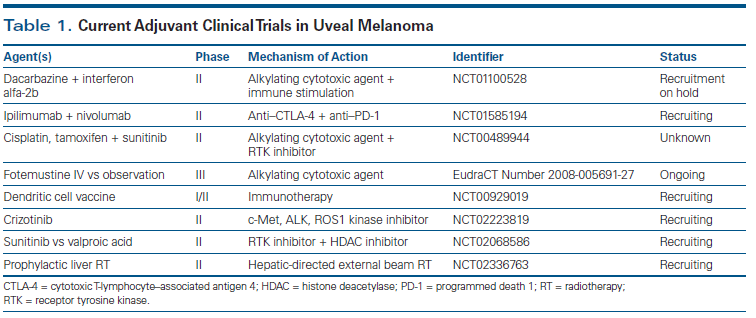
Significant effort has been made to better elucidate the biological mechanisms driving distant spread and growth. A number of growth factor receptors, such as c-Met, c-Kit, and insulin growth factor receptor 1, have been implicated in the metastatic progression of uveal melanoma.[44-46] Inhibitors of these receptor tyrosine kinases, which include crizotinib and sunitinib, are currently being tested as adjuvant therapies (Table 1). The protein MDA-9/syntenin-1 (melanoma differentiation associated gene 9/syndecan-binding protein 1), encoded by the SDCBP gene, may also be involved in metastatic dissemination via increased hepatocellular growth factor–mediated invasion (activation of focal adhesion kinase [FAK], Akt, Src).[47] The liver is the predominant site of metastasis, seen in over 90% of cases, possibly because of differential expression of genes involved in tumor cell proliferation, adhesion, and migration/invasion (eg, HTR2B, CHL1, ZNF, YWHAZ, FYN).[48] The chemokine receptor CXCR4 (C-X-C chemokine receptor type 4) has also been shown to play a role in the liver homing of uveal melanoma cells.[49] Lung, bone, and skin are other common sites for metastasis.[50,51] In addition, loss of the tumor suppression gene encoding BAP1 is highly associated with the metastatic potential of uveal melanoma.[9,25] Histone deacetylases, including valproic acid, have been found to counteract the effect of BAP1 loss and to promote uveal melanoma differentiation in animal models; valproic acid is currently being tested as an adjuvant therapy (see Table 1).[52]
Systemic treatment in the adjuvant setting could possibly prevent or delay the development of macrometastases, which are difficult to treat once they are established. However, no systemic adjuvant therapy thus far has been shown to reduce the risk of metastasis. A randomized trial in 1998 did not find a statistically significant difference in the 5-year survival rate with dacarbazine treatment (71%) compared with observation (68%).[53] Based on data showing a benefit from adjuvant interferon in cutaneous melanoma, another trial evaluated the efficacy of adjuvant maintenance interferon alfa-2a following primary treatment with either enucleation or proton beam radiation. There was no survival benefit with interferon therapy compared with matched historical controls in this study.[54]
Current approaches are targeting uveal melanoma more rationally, as well as using molecular analysis for a more uniform selection of high-risk patients. Given the poor prognosis of these patients, clinical trial participation should be considered in patients who are at high risk for recurrent disease. Table 1 lists therapies currently being investigated in an adjuvant setting. These strategies include receptor tyrosine kinase inhibition, histone deacetylase inhibition for BAP1 loss, and liver-directed therapies. Immune-modulating approaches are also being evaluated in the adjuvant setting; these include anti–cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) and anti–programmed death 1 (PD-1) treatments, in addition to the development of a vaccine consisting of autologous dendritic cells electroporated with the melanoma antigens gp100 and tyrosinase, for use in patients with high-risk Class 2 uveal melanoma (ClinicalTrials.gov identifier: NCT00929019; see Table 1).
Treatment of metastatic uveal melanoma
The long-term prognosis of metastatic disease is very poor. The median survival time from the development of distant metastases is between 4 and 15 months, and the 1-year survival rate is 10% to 15%.[28,50,55] There is currently no standard-of-care therapy for metastatic uveal melanoma. A broad range of treatments has been evaluated, including chemotherapy, immunotherapy, and molecularly targeted agents. Hepatic-directed treatments have also been developed.
Systemic chemotherapy. Systemic chemotherapeutic regimens in uveal melanoma have been adopted from those used in cutaneous melanoma, including dacarbazine-, temozolomide-, cisplatin-, treosulfan-, and fotemustine-based regimens. To date, no conventional chemotherapeutic agent has been found to extend survival in patients with metastatic disease, and the response rates range from 0% to 15%.[56-61]
Systemic immunotherapy. Recent advances in immunotherapy have dramatically improved survival for patients with metastatic cutaneous melanoma. However, the clinical benefit in uveal melanoma thus far has been more limited. One possible explanation for the difference in response is that uveal melanoma is characterized by a low mutational burden, since UV radiation–induced DNA damage does not appear to play a significant role in tumor pathogenesis.[62] High mutational load caused by chronic mutagen exposure is thought to generate neoantigens that can elicit an antitumor response that is augmented by immune checkpoint inhibition. In cutaneous melanoma and non–small-cell lung cancer, the clinical response to anti–CTLA-4 and anti–PD-1 therapy has been found to correlate with elevated mutational and neoantigen burden.[63,64] In primary uveal melanoma, another possible explanation derives from the concept of “immune privilege,” an adaptation to reduce immune-mediated injury in organs such as the eye and brain that have limited capacity for regeneration. Studies have shown that aqueous humor contains a number of anti-inflammatory and immunosuppressive cytokines, such as macrophage migration inhibitory factor and transforming growth factor β. Iris and ciliary body cells directly suppress T-cell proliferation by induction of regulatory T-cell activity. Endothelial cells lining the anterior chamber downregulate major histocompatibility complex class I expression and secrete indoleamine 2,3-dioxygenase (IDO), an enzyme that deprives T cells of tryptophan and has immunosuppressive effects on the tumor microenvironment.[65,66]
The immune modulating agent ipilimumab, a humanized monoclonal antibody against CTLA-4, has shown clinical benefit in patients with metastatic cutaneous melanoma.[67] However, the efficacy of ipilimumab in treating uveal melanoma is unclear. Overall response rates in metastatic uveal melanoma are 5% to 10%, with a median OS of 6.0 to 9.7 months.[68-71] In the phase II GEM-1 trial evaluating ipilimumab, 10 mg/kg, in treatment-naive patients with metastatic uveal melanoma, preliminary results suggest promising clinical activity (7.7% partial response rate and 46.2% stable disease rate after a 5.5-month median follow-up); however, final results are still pending.[72] The phase II DeCOG trial, in which both treatment-naive and pretreated patients received ipilimumab, 3 mg/kg, found limited clinical benefit (progression-free survival [PFS] of 2.8 months, median OS of 6.8 months). However, the treatment-naive cohort was small and received a lower dose than patients in the GEM-1 trial.[73]
The anti–PD-1 antibodies nivolumab and pembrolizumab have been approved by the US Food and Drug Administration (FDA) for the treatment of metastatic cutaneous melanoma, and generally have a more tolerable side-effect profile than ipilimumab.[74-76] Under an expanded access program, seven patients with metastatic uveal melanoma received pembrolizumab after their disease had progressed on ipilimumab. Initial results showed a median PFS of 12.2 weeks.[77] Further investigation is needed, and a phase II study of pembrolizumab in advanced uveal melanoma has been initiated (NCT02359851). Combination treatment with ipilimumab and nivolumab is also being assessed in a phase II trial (NCT01585194).
In addition, phase II investigation of autologous tumor-infiltrating lymphocytes administered with or without high doses of interleukin-2 following chemotherapy in patients with metastatic disease is underway (NCT01814046).
Molecularly targeted therapy. Several promising therapeutic targets have been identified as a result of our increased understanding of the oncogenic drivers in uveal melanoma. Because the MAPK pathway is activated in the majority of uveal melanoma cases, inhibitors of downstream effectors, such as MEK and protein kinase C (PKC), are currently under clinical investigation. Of note, the common BRAF mutations found in cutaneous melanoma are present in less than 1% of uveal melanoma tumors, rendering BRAF inhibitors unsuitable for treating uveal melanoma.
MEK inhibition with selumetinib showed promising clinical efficacy in a randomized phase II trial of selumetinib vs chemotherapy. The selumetinib-treated group had improved PFS and response rate compared with either temozolomide or dacarbazine chemotherapy in both pretreated and treatment-naive patients with metastatic uveal melanoma. The objective response rate was 14% with selumetinib, compared with 0% in the chemotherapy-treated arm, and the median PFS was 15.9 weeks and 7 weeks, respectively.[78] Patients with wild-type exon 5 of GNAQ and GNA11 had a longer PFS than those carrying exon 5 mutations (25.9 weeks [range, 3.7 to 40.4 weeks] vs 15.4 weeks [range, 8.1 to 16.8 weeks]). However, the wild-type exon 5 cohort may have harbored activating mutations in exon 4 of GNAQ or GNA11, which exist in a mutually exclusive pattern vis à vis exon 5 mutations; exon 4 mutations were not routinely genotyped in the study, since these had not been described at the time of the study’s initiation. No OS benefit was observed, but the survival data may have been confounded by the design of the study.
In contrast, the recently completed multicenter phase III SUMIT study comparing selumetinib plus dacarbazine vs dacarbazine alone did not show a statistically significant improvement in PFS, possibly because of differences in the patient population and study design from those of the phase II trial.[79] The phase II trial utilized a monotherapy investigational study arm, while the phase III trial evaluated the combination of selumetinib and dacarbazine. Moreover, there were discrepancies in results reported by the central and site-based radiologists. According to blinded independent central radiology review of PFS, the SUMIT study’s primary endpoint, selumetinib plus dacarbazine resulted in a median PFS of 2.8 months, compared with 1.8 months with placebo plus dacarbazine (hazard ratio [HR], 0.78 [95% confidence interval (CI), 0.48–1.27]; 2-sided P = .3195). Objective response rate by central review was 3.1% and 0% (P = .36) for selumetinib plus dacarbazine and placebo plus dacarbazine, respectively. According to site-based radiology review, however, a numerical difference was observed in PFS between the two groups, with a median PFS of 3.8 months in the selumetinib-plus-dacarbazine group vs 2.1 months in the placebo-plus-dacarbazine group (HR, 0.49 [95% CI, 0.28–0.84]).[79] Further investigation is ongoing to evaluate these differences in radiology review.
The receptor tyrosine kinase c-Kit has been identified as a potential oncogenic driver in uveal melanoma, based on immunohistochemistry analysis of resected tumors.[45] However, the SUAVE trial, a phase II trial comparing sunitinib, a nonselective c-Kit inhibitor, with dacarbazine showed no improvement in PFS or OS with sunitinib.[80]
Preclinical studies have identified c-Met, the receptor tyrosine kinase for the ligand hepatocyte growth factor, as another potential target in uveal melanoma; c-Met is overexpressed in uveal melanoma, and inhibition of c-Met was shown to prevent tumor growth in preclinical models of uveal melanoma.[81,82] Reanalysis of a discontinued phase II trial, which encompassed all melanomas, revealed that the small-molecule drug used in that trial, cabozantinib, displayed activity in the subset of patients with uveal melanoma. Of the patients whose tumor genotype was determined, 90% harbored GNAQ or GNA11 mutations.[83] Based on these results, a new phase II trial is currently underway for the treatment of metastatic uveal melanoma with cabozantinib (NCT01835145).
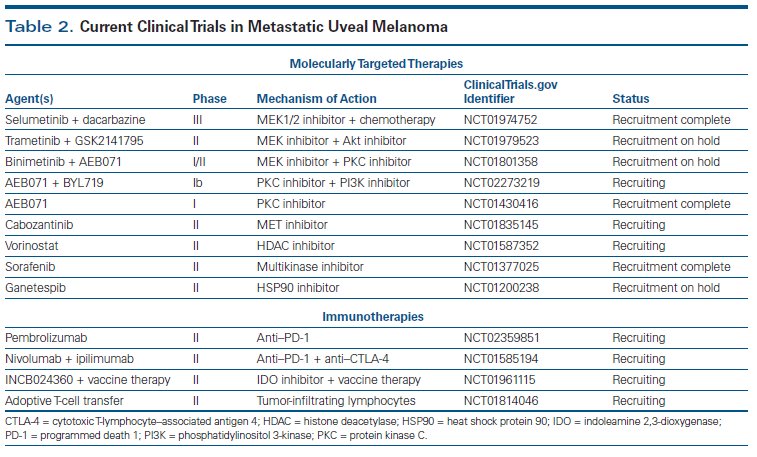
A number of other trials for patients with metastatic uveal melanoma are ongoing (see Table 2).
Liver-directed therapies
In highly selected cases, surgical removal of metastatic nodules can provide long-term survival benefit, and in a very few patients may be curative.[84] Other locoregional modalities, such as radiofrequency ablation, cryotherapy, and stereotactic radiotherapy, are possible alternative approaches.[85]
Other liver-directed therapies take advantage of the liver’s dual blood supply in order to deliver treatments more directly to the metastases through the hepatic artery. Recruited hepatic artery branches vascularize the melanoma, whereas the portal circulation provides the majority of blood to the normal liver tissue. Intrahepatic therapeutic approaches include bland embolization, intra-arterial administration of chemotherapies, isolated hepatic perfusion, intra-arterial hepatic chemoembolization, and immunoembolization.[84,86,87]
Two randomized phase III trials of liver-directed therapies have been completed to date. In one trial, improved response rate and PFS were observed in patients receiving hepatic intra-arterial fotemustine compared with intravenous administration; however, no OS benefit was observed.[88] The other phase III trial tested the nonsurgical technique of percutaneous hepatic perfusion of melphalan vs best alternative care. Improved hepatic PFS and overall response rate were observed in the percutaneous hepatic perfusion group; however, no improvement in OS was observed. The lack of an OS benefit could be due to the crossover study design, and another phase III trial is being initiated.[89]
Conjunctival Melanoma
TO PUT THAT INTO CONTEXT
[[{"type":"media","view_mode":"media_crop","fid":"44935","attributes":{"alt":"","class":"media-image","id":"media_crop_9744963451770","media_crop_h":"0","media_crop_image_style":"-1","media_crop_instance":"5073","media_crop_rotate":"0","media_crop_scale_h":"0","media_crop_scale_w":"0","media_crop_w":"0","media_crop_x":"0","media_crop_y":"0","style":"height: 180px; width: 155px;","title":" ","typeof":"foaf:Image"}}]]
Ahmad Tarhini, MD, PhD
University of Pittsburgh Cancer Institute
Pittsburgh, PennsylvaniaWhat Do We Know About the Biology of Uveal Melanoma?Unlike cutaneous melanomas, in which driver mutations are mainly associated with the mitogen-activated protein kinase (MAPK) pathway (eg, BRAF, NRAS, NF1), uveal melanomas appear to be driven by point mutations in the G protein α-subunits (GNAQ or GNA11). These mutations, found in about 80% of cases, lead to activation of the MAPK and other pathways.What Are the Key Prognostic Factors in Uveal Melanoma?
Clinically, prognosis has been associated with patient age and characteristics of the primary tumor (tumor size, extrascleral extension, involvement of the ciliary body, and incomplete local control). The TNM staging system has also been shown to be prognostic and has been validated. Further, a gene expression profile consisting of a 15-gene signature was shown to improve risk stratification as compared with the TNM staging system. Certain genetic alterations also have prognostic value.What Are the Main Treatment Options for Localized Uveal Melanoma?
In the United States, plaque brachytherapy is the most frequently used treatment for localized uveal melanoma. Other options include charged-particle radiotherapy, proton beam therapy, and surgery. With a primary tumor, such local treatment modalities are effective at preventing local recurrence in over 95% of cases. However, they have no impact on the risk of distant relapse.What Are the Systemic Therapeutic Options for Patients With Uveal Melanoma?
To date, adjuvant therapy after the initial management of a primary uveal melanoma has not been shown to affect the risk of relapse or death. Therefore, close observation is the standard of care. For patients with metastatic uveal melanoma, the prognosis is poor, with a median survival of 4 to 15 months and a 1-year survival rate of 10% to 15%. Ongoing clinical trials are promising, however; driven by significant advances in our understanding of the biology of uveal melanoma, these involve chemotherapeutic, immunotherapeutic, and molecularly targeted agents.Financial Disclosure:Dr. Tarhini serves as a consultant to Bristol-Myers Squibb and Merck; he has done contracted research for Amgen, Bristol-Myers Squibb, Incyte, Merck, and Novartis.
Epidemiology and risk factors
Conjunctival melanoma is a rare ocular tumor comprising about 5% of ocular melanomas and 0.25% of all melanomas.[1,90] Conjunctival melanoma arises from melanocytes among the basal cells of the conjunctival epithelium. The incidence of conjunctival melanoma in the United States has risen over the past few decades, consistent with the rising incidence observed in cutaneous melanoma cases. A SEER database analysis found that the incidence of conjunctival melanoma cases increased by 101% from 1973 to 1999, with the greatest change seen among white men.[91] In Sweden, the age-adjusted incidence of conjunctival melanoma increased by sevenfold between 1960 and 2005, and in Finland it increased twofold between 1967 and 2000.[92,93]
Risk factors for conjunctival melanoma have not been well defined, in part because of the small number of cases. In the United States, conjunctival melanoma is more common in Caucasians than in African-Americans (2.6:1 incidence ratio).[94] Around 74% of cases arise from primary acquired melanosis (PAM), 7% arise from a pre-existing nevus, and about 19% arise de novo.[95]
Molecular biology
The molecular pathogenesis of conjunctival melanoma is distinct from that of uveal melanoma. Activation of the MAPK pathway via BRAF and NRAS mutations has been reported in a subset of conjunctival melanoma cases. A case series of 78 patients with conjunctival melanoma reported BRAF V600E mutations in 27% of tumors and NRAS mutations in 18%.[96] KIT mutations are found in subsets of cutaneous and mucosal melanomas, and have also been reported as accounting for 0% to 7% of conjunctival melanoma cases.[97,98] Activation of the mammalian target of rapamycin (mTOR) pathway, confirmed by high expression of phosphorylated AKT, S6, and p-4E-BP1, has also been reported in conjunctival melanoma.[99] Other genetic patterns include amplification of CDKN1A and RUNX2 in primary tumors. Metastatic lesions demonstrate amplification of MLH1 and TIMP2 and deletion of MGMT and ECHS1.[100]
Surgical treatment of conjunctival melanoma
The rarity of conjunctival melanoma has resulted in a paucity of data to guide management. Treatment options are largely based on a small number of case series. The current standard practice for conjunctival melanoma involves surgical excision and may include adjuvant therapy.
Surgical resection. Radical techniques, such as enucleation and orbital exenteration, have been used for treatment of extensive conjunctival melanoma, but no data demonstrate that this practice improves survival, and it is associated with significant morbidity.[101,102] Thus, these techniques are currently used only as palliation for highly invasive tumors. The current standard surgical approach is wide local excision and biopsy to minimize seeding, tumor dissemination, and recurrence.[103,104] Excision is typically followed by cryotherapy to the margins. In cases of scleral or corneal invasion, partial sclerectomy or alcohol epitheliectomy to the cornea are also performed.[103]
Sentinel lymph node biopsy. Although sentinel lymph node biopsy (SLNB) has an established role in the management of cutaneous melanoma, its use in this setting remains controversial. Roughly 50% of patients with cutaneous melanoma may have metastatic spread to regional lymph nodes (preauricular, submandibular, deep cervical) before systemic involvement develops.[105,106] Proponents of SLNB believe that, by detecting subclinical micrometastasis to regional nodes, SLNB can guide early disease treatment before the development of distant or systemic metastases. However, up to 50% of patients, particularly those with prior local recurrence, can develop distant metastasis without evidence of regional lymph node involvement.[105,106] Moreover, there remains a lack of consensus as to when such an invasive procedure is more likely to yield positive findings. Reported risk factors for regional spread include tumor thickness > 2 mm, tumor width > 10 mm, positive surgical margins, nonlimbal location, and the presence of histologic ulceration.[107-109] Even with appropriate patient selection, the positivity rate for SLNB in conjunctival melanoma appears to be low (11% to 15%) based on two case series involving a total of 44 patients.[109,110] Of note, in the study performed by Savar and colleagues, three out of four patients with conjunctival melanoma who were found to have positive lymph nodes and who subsequently underwent complete neck dissection eventually developed local recurrence and widespread metastatic disease.[109] Larger trials will be needed to determine uniform protocols for SLNB and whether routine SLNB leads to improvement in survival.
Adjuvant therapy
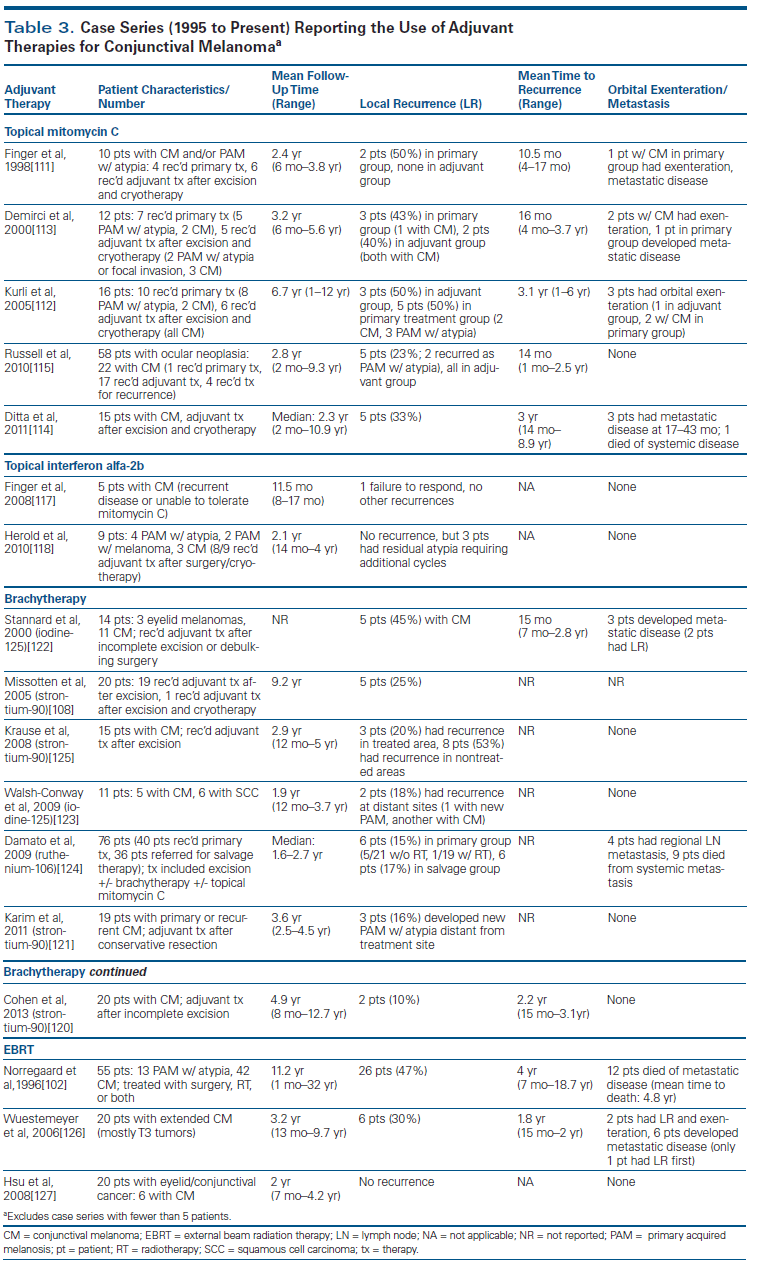
The use of local adjuvant therapy may improve long-term control and prevent local recurrence following surgery, particularly in the case of lesions that cannot be fully excised, where tumor is present at the surgical margins. Given the absence of large clinical trials, there is no consensus on the preferred adjuvant agent. Table 3 summarizes reported case series of adjuvant therapy use to date. Available options include cryotherapy, as previously discussed, topical chemotherapy, and various radiotherapies.
Topical chemotherapy. Of the topical agents, mitomycin C is the most studied and most commonly used agent. It is an established treatment for PAM with atypia, but its role in the management of conjunctival melanoma is limited to the adjuvant setting, given the relatively poor outcomes when it is used as primary therapy. The majority of patients who received topical mitomycin C as primary treatment in prior studies developed recurrent disease requiring exenteration.[111-113]
As adjuvant therapy, mitomycin C is given 2 weeks after surgical excision to allow wound healing. A review of several case series found recurrence rates ranging from 0% to 50% after mean follow-up times of 2.4 to 6.7 years.[111-115] Treatment efficacy was largely dependent on the depth of the lesion, since mitomycin C does not cross the basement membrane. Multifocal and nodular disease were more resistant to treatment, and recurrences tended to involve deeper layers of the lamina propria, with sparing of the epithelium and superficial layers. Common adverse effects of treatment with mitomycin C have included allergic reaction and local irritation.[114,116]
Interferon alfa-2b can be applied topically or given as subconjunctival/perilesional injections. Topical application is generally preferred because it results in fewer side effects and avoids systemic toxicity. Data on the use of interferon for conjunctival melanoma is limited to two case series involving ten patients.[117,118] There were no local recurrences (one patient failed to respond), but the follow-up time (8 to 27 months) was generally shorter than that in the mitomycin C studies. Given its minimal toxicity, interferon may be considered a reasonable alternative for patients who cannot tolerate topical mitomycin C.
Radiotherapies. Radiotherapy can be administered as external beam radiation therapy (EBRT) or local brachytherapy; the latter is generally preferred, since it minimizes radiation to the rest of the ocular surface. Treatment is delayed following surgical excision to allow the conjunctiva to heal. Brachytherapy is delivered via plaques, applicators, or interstitial implants using a number of different isotopes. The type of radiotherapy and route of delivery are generally chosen based on the location and size of the tumor.[119] Outcomes for adjuvant brachytherapy following excision were evaluated in seven studies, four of which used strontium-90.[108,120-125] The rate of local recurrence, both at treated and untreated sites, ranged from 10% to 53%. Mean follow-up time ranged from 1.9 to 9.2 years. Brachytherapy is generally well tolerated, with mostly transient side effects. Common adverse effects include dry eye syndrome and corneal ulcers.
Data on EBRT are more limited, given its minimal use compared with brachytherapy. Outcomes appear poorer than those seen with brachytherapy, with 30% to 47% of patients experiencing local recurrence and up to 30% of patients developing metastatic disease during the follow-up period (2.0 to 11.2 years). However, studies using EBRT have included patients with more aggressive and extensive tumors. As expected, EBRT is associated with a higher frequency of more severe adverse effects because of the more widespread exposure to radiation.[102,126,127]
Screening and surveillance
There are no clear guidelines regarding surveillance for metastatic disease in patients following treatment of a primary conjunctival melanoma. However, it would be reasonable to consider routine imaging studies for all patients with high-risk features.
Treatment of metastatic disease
Given the rarity of conjunctival melanoma, there is no standard recommendation for the treatment of patients with metastatic disease. In general, these patients are currently treated in a manner similar to that used in patients with advanced cutaneous melanoma. A number of genetic mutations have been identified in conjunctival melanoma, creating the opportunity for biologic therapies that target these specific mutations. Of these mutations, BRAF has received the most attention, given the established role of BRAF inhibitors in cutaneous melanoma treatment. Data from various cohorts show that up to 50% of primary and metastatic conjunctival melanomas harbor a mutation in BRAF, the majority of which are V600E mutations.[96,100,128] No clinical trials have yet assessed the efficacy of BRAF inhibitors (vemurafenib, dabrafenib) in conjunctival melanoma. However, it would be reasonable to routinely test for the BRAF V600E mutation; test results could guide the choice of treatment for patients who develop systemic metastatic disease. The mTOR pathway may also be overactivated in conjunctival melanoma, suggesting a possible role for mTOR inhibitors such as rapamycin.[99]
Prognosis
The rate of local recurrence is reported to be 30% to 50% at 5 years.[108] Risk factors for recurrence include multifocal disease, increased tumor thickness, incomplete excision with positive margins, surgical excision without adjuvant therapy, and nonlimbal location.[93,107] In particular, patients with tumors located in the forniceal conjunctiva, caruncle, plica semilunaris, or eyelid margin have been shown to have higher rates of recurrence and a poorer prognosis. Conversely, epibulbar tumors have been associated with lower rates of recurrence and metastasis.[108,124] Tumor location, along with poor visual acuity and melanoma color (red), have also been predictive of the need for orbital exenteration.[108,129] Roughly 30% of patients may require eventual exenteration for refractory disease.[108,129]
Metastatic disease develops in around 20% to 30% of patients within 10 years.[104,105,128] Local recurrence itself contributes to the risk for metastasis and increased mortality. Other poor prognostic clinical features include de novo lesions; tumor thickness > 2 mm; nodular growth pattern; and involvement of nonbulbar conjunctiva, medial bulbar conjunctiva, caruncle, or plica semilunaris. Distant metastasis most commonly involves the lung, brain, liver, skin, bone, and gastrointestinal tract.[95,106-108,130] Estimated 5- and 10-year mortality rates are ~10%–20% and ~20%–40%, respectively.[93,107,108,129,131]
Future Directions
Uveal melanoma
Currently, there is no standard-of-care treatment for metastatic uveal melanoma and no therapy that has been proven to improve OS. Despite the results observed with combined selumetinib and dacarbazine in the SUMIT trial, MEK inhibition may still have a role in the treatment of metastatic uveal melanoma. Future strategies to optimize the efficacy of MEK inhibition in uveal melanoma should include targeting multiple nodes within the MAPK pathway, or targeting parallel pathways (such as the PI3K/Akt pathway). In preclinical studies, combined inhibition of MEK and PKC resulted in MAPK pathway inhibition in cell lines and tumor regression in a xenograft mouse model.[132] The combination of PKC and MEK inhibition with AEB071 and binimetinib was evaluated in a phase Ib/II trial (NCT01801358), but the results have not yet been reported. Given that multiple pathways are activated in the development of uveal melanoma, targeting parallel pathways, such as the MAPK and PI3K/Akt pathways, may also improve clinical efficacy. Because preclinical in vitro studies and xenograft mouse models have shown synergy when MEK and Akt inhibition are combined,[133] there is an ongoing randomized phase II trial of MEK inhibition with trametinib with or without Akt inhibition with GSK2141795 in metastatic uveal melanoma (NCT01979523). Lastly, exploring alternate dosing schedules may enhance the effect of MEK inhibition by delaying the onset of drug resistance and enhancing the durability of responses. Preclinical studies evaluating an intermittent dosing schedule of vemurafenib in BRAF-mutated cutaneous melanoma resulted in delayed onset of drug resistance and temporary tumor regression.[134] Further investigation of an intermittent dosing schedule with MEK inhibitors in uveal melanoma should be considered.
Recent work by Yu et al and Feng et al identified the YAP pathway as another potential therapeutic target in uveal melanoma.[14,15] Verteporfin, a derivative of the porphyrin family that is currently FDA-approved for use in photodynamic therapy to eliminate neovascularization of blood vessels in the eye, was shown to inhibit YAP activity in these recent preclinical studies in uveal melanoma; in addition, it resulted in reduced proliferation of uveal melanoma cells in vitro and reduced tumor growth in vivo.[14,15] These results suggest that pharmacologic inhibition of YAP may represent another potential therapeutic strategy in uveal melanoma.
Efforts to further the understanding of the biology of uveal melanoma are crucial to the development of new therapeutic strategies in this disease. The TCGA program recently characterized the genomic alterations of 333 primary or metastatic cutaneous melanoma samples,[10] and genomic analysis of uveal melanoma is currently ongoing as part of the Rare Tumor Projects of the TCGA program. The results of this analysis will hopefully lead to identification of new therapeutic targets in this rare subtype of melanoma.
Lastly, immunotherapy in uveal melanoma remains an area of active exploration. While checkpoint inhibition with anti–PD-1 and anti–CTLA-4 therapy has drastically changed the treatment approach to cutaneous melanoma, its efficacy in uveal melanoma is still being evaluated. Although the clinical benefit of single-agent ipilimumab in phase II trials was variable,[68-73] current clinical trials are ongoing to evaluate the efficacy of PD-1 blockade with pembrolizumab and combined CTLA-4 and PD-1 blockade with ipilimumab and nivolumab. Adoptive T-cell transfer is currently being evaluated in an ongoing phase II study (NCT01814046), and vaccine therapy is being evaluated in combination with IDO inhibition (NCT01961115; see Table 2).
Conjunctival melanoma
Given that conjunctival melanoma is extremely rare, the biology and the potential of targeted therapies and immunotherapies in this subtype of ocular melanoma have not been well characterized thus far. In light of the frequency of BRAF mutations in conjunctival melanoma, further exploration of BRAF and MEK inhibitor therapy is warranted in metastatic disease. Ongoing efforts to better characterize the genomic alterations in conjunctival melanoma may help identify future treatment strategies.
Conclusion
Ocular melanoma is a rare but diverse disease that includes uveal, conjunctival, and orbital melanoma. The biologies of uveal and conjunctival melanoma are distinct from each other-as well as from that of cutaneous melanoma-and thus require different treatment strategies. While there is no standardized treatment for either metastatic uveal or metastatic conjunctival melanoma, significant advances have been made in our understanding of the biology of these rare melanoma subtypes, leading to novel targeted therapy and immunotherapy approaches. Further studies are needed to understand and improve the efficacy of targeted therapy and immunotherapy in ocular melanoma.
Financial Disclosure: Dr. Carvajal has served as a consultant to AstraZeneca, Janssen Pharmaceutica, Merck, and Novartis. Drs. Blum, Yang, and Komatsubara have no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
References:
1. McLaughlin CC, Wu XC, Jemal A, et al. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005;103:1000-7.
2. Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881-5.
3. Virgili G, Gatta G, Ciccolallo L, et al. Incidence of uveal melanoma in Europe. Ophthalmology. 2007;114:2309-15.
4. Hu DN, Yu GP, McCormick SA, et al. Population-based incidence of uveal melanoma in various races and ethnic groups. Am J Ophthalmol. 2005;140:612-7.
5. Shah CP, Weis E, Lajous M, et al. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112:1599-607.
6. Mainster MA, Turner PL. Ultraviolet-B phototoxicity and hypothetical photomelanomagenesis: intraocular and crystalline lens photoprotection. Am J Ophthalmol. 2010;149:543-9.
7. Shields CL, Kaliki S, Livesey M, et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: analysis of 7872 consecutive eyes. JAMA Ophthalmol. 2013;131:993-1003.
8. Weis E, Shah CP, Lajous M, et al. The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol. 2006;124:54-60.
9. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410-3.
10. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681-96.
11. Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599-602.
12. Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191-9.
13. Shoushtari AN, Carvajal RD. GNAQ and GNA11 mutations in uveal melanoma. Melanoma Res. 2014;24:525-34.
14. Yu FX, Luo J, Mo JS, et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822-30.
15. Feng X, Degese MS, Iglesias-Bartolome R, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831-45.
16. Whitehead J, Tishkovskaya S, O’Connor J, Damato B. Devising two-stage and multistage phase II studies on systemic adjuvant therapy for uveal melanoma. Invest Ophthalmol Vis Sci. 2012;53:4986-9.
17. Shields CL, Furuta M, Thangappan A, et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009;127:989-98.
18. Egger E, Zografos L, Schalenbourg A, et al. Eye retention after proton beam radiotherapy for uveal melanoma. Int J Radiat Oncol Biol Phys. 2003;55:867-80.
19. Schmittel A, Bechrakis NE, Martus P, et al. Independent prognostic factors for distant metastases and survival in patients with primary uveal melanoma. Eur J Cancer. 2004;40:2389-95.
20. Li W, Gragoudas ES, Egan KM. Metastatic melanoma death rates by anatomic site after proton beam irradiation for uveal melanoma. Arch Ophthalmol. 2000;118:1066-70.
21. Shields CL, Kaliki S, Furuta M, et al. American Joint Committee on Cancer classification of uveal melanoma (anatomic stage) predicts prognosis in 7,731 patients: the 2013 Zimmerman Lecture. Ophthalmology. 2015;122:1180-6.
22. Harbour JW, Chen R. The DecisionDx-UM gene expression profile test provides risk stratification and individualized patient care in uveal melanoma. PLoS Curr. 2013 Apr 9;5.
23. Onken MD, Worley LA, Char DH, et al. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012;119:1596-603.
24. Augsburger JJ, Correa ZM, Trichopoulos N. Surveillance testing for metastasis from primary uveal melanoma and effect on patient survival. Am J Ophthalmol. 2011;152:5-9 e1.
25. Gupta MP, Lane AM, DeAngelis MM, et al. Clinical characteristics of uveal melanoma in patients with germline BAP1 mutations. JAMA Ophthalmol. 2015;133:881-7.
26. Harbour JW, Roberson ED, Anbunathan H, et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133-5.
27. Martin M, Masshofer L, Temming P, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933-6.
28. Nathan P, Cohen V, Coupland S, et al. Uveal melanoma UK national guidelines. Eur J Cancer. 2015;51:2404-12.
29. Collaborative Ocular Melanoma Study G. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol. 2006;124:1684-93.
30. Wang Z, Nabhan M, Schild SE, et al. Charged particle radiation therapy for uveal melanoma: a systematic review and meta-analysis. Int J Radiat Oncol Biol Phys. 2013;86:18-26.
31. Gragoudas E, Li W, Goitein M, et al. Evidence-based estimates of outcome in patients irradiated for intraocular melanoma. Arch Ophthalmol. 2002;120:1665-71.
32. Damato B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye (Lond). 2012;26:1157-72.
33. Tarmann L, Wackernagel W, Avian A, et al. Ruthenium-106 plaque brachytherapy for uveal melanoma. Br J Ophthalmol. 2015 May 15. [Epub ahead of print]
34. Mishra KK, Quivey JM, Daftari IK, et al. Long-term results of the UCSF-LBNL randomized trial: charged particle with helium ion versus iodine-125 plaque therapy for choroidal and ciliary body melanoma. Int J Radiat Oncol Biol Phys. 2015;92:376-83.
35. Yarovoy AA, Magaramov DA, Bulgakova ES. The comparison of ruthenium brachytherapy and simultaneous transpupillary thermotherapy of choroidal melanoma with brachytherapy alone. Brachytherapy. 2012;11:224-9.
36. Sagoo MS, Shields CL, Mashayekhi A, et al. Plaque radiotherapy for juxtapapillary choroidal melanoma: tumor control in 650 consecutive cases. Ophthalmology. 2011;118:402-7.
37. Hawkins BS; Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol. 2004;138:936-51.
38. Melia M, Moy CS, Reynolds SM, et al. Quality of life after iodine 125 brachytherapy vs enucleation for choroidal melanoma: 5-year results from the Collaborative Ocular Melanoma Study: COMS QOLS Report No. 3. Arch Ophthalmol. 2006;124:226-38.
39. Bechrakis NE, Petousis V, Willerding G, et al. Ten-year results of transscleral resection of large uveal melanomas: local tumour control and metastatic rate. Br J Ophthalmol. 2010;94:460-6.
40. Willerding GD, Cordini D, Moser L, et al. Neoadjuvant proton beam irradiation followed by transscleral resection of uveal melanoma in 106 cases. Br J Ophthalmol. 2015 July 29. [Epub ahead of print]
41. Torres V, Triozzi P, Eng C, et al. Circulating tumor cells in uveal melanoma. Future Oncol. 2011;7:101-9.
42. Clarijs R, Schalkwijk L, Ruiter DJ, de Waal RM. Lack of lymphangiogenesis despite coexpression of VEGF-C and its receptor Flt-4 in uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42:1422-8.
43. Refaian N, Schlereth SL, Koch KR, et al. Comparing the hem- and lymphangiogenic profile of conjunctival and uveal melanoma cell lines. Invest Ophthalmol Vis Sci. 2015;56:5691-7.
44. Mallikarjuna K, Pushparaj V, Biswas J, Krishnakumar S. Expression of epidermal growth factor receptor, ezrin, hepatocyte growth factor, and c-Met in uveal melanoma: an immunohistochemical study. Curr Eye Res. 2007;32:281-90.
45. All-Ericsson C, Girnita L, Muller-Brunotte A, et al. c-Kit-dependent growth of uveal melanoma cells: a potential therapeutic target? Invest Ophthalmol Vis Sci. 2004;45:2075-82.
46. All-Ericsson C, Girnita L, Seregard S, et al. Insulin-like growth factor-1 receptor in uveal melanoma: a predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol Vis Sci. 2002;43:1-8.
47. Gangemi R, Mirisola V, Barisione G, et al. Mda-9/syntenin is expressed in uveal melanoma and correlates with metastatic progression. PLoS One. 2012;7:e29989.
48. Zhang Y, Yang Y, Chen L, Zhang J. Expression analysis of genes and pathways associated with liver metastases of the uveal melanoma. BMC Med Genet. 2014;15:29.
49. Li H, Yang W, Chen PW, et al. Inhibition of chemokine receptor expression on uveal melanomas by CXCR4 siRNA and its effect on uveal melanoma liver metastases. Invest Ophthalmol Vis Sci. 2009;50:5522-8.
50. Diener-West M, Reynolds SM, Agugliaro DJ, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123:1639-43.
51. Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch Ophthalmol. 2001;119:670-6.
52. Landreville S, Agapova OA, Matatall KA, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18:408-16.
53. Desjardins L, Dorval T, Levy C, et al. Randomised study on adjuvant therapy by DTIC in choroidal melanoma. Ophtalmogie. 1998;12:168-73.
54. Lane AM, Egan KM, Harmon D, et al. Adjuvant interferon therapy for patients with uveal melanoma at high risk of metastasis. Ophthalmology. 2009;116:2206-12.
55. Postow MA, Kuk D, Bogatch K, Carvajal RD. Assessment of overall survival from time of metastasis in mucosal, uveal, and cutaneous melanoma. J Clin Oncol. 2014;32(suppl 5s):abstr 9074.
56. Spagnolo F, Caltabiano G, Queirolo P. Uveal melanoma. Cancer Treat Rev. 2012;38:549-53.
57. Spagnolo F, Grosso M, Picasso V, et al. Treatment of metastatic uveal melanoma with intravenous fotemustine. Melanoma Res. 2013;23:196-8.
58. Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009;148:119-27.
59. Pereira PR, Odashiro AN, Lim LA, et al. Current and emerging treatment options for uveal melanoma. Clin Ophthalmol. 2013;7:1669-82.
60. Schmittel A, Schmidt-Hieber M, Martus P, et al. A randomized phase II trial of gemcitabine plus treosulfan versus treosulfan alone in patients with metastatic uveal melanoma. Ann Oncol. 2006;17:1826-9.
61. Homsi J, Bedikian AY, Papadopoulos NE, et al. Phase 2 open-label study of weekly docosahexaenoic acid-paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010;20:507-10.
62. Furney SJ, Pedersen M, Gentien D, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122-9.
63. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189-99.
64. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8.
65. Niederkorn JY. Ocular immune privilege and ocular melanoma: parallel universes or immunological plagiarism? Front Immunol. 2012;3:148.
66. de Waard-Siebinga I, Hilders CG, Hansen BE, et al. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 1996;234:34-42.
67. Dummer R, Hauschild A, Guggenheim M, et al. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7):vii86-vii91.
68. Deo MA. Long-term survival benefit from ipilimumab treatment in metastatic uveal melanoma patients. J Clin Oncol. 2014;32(suppl 5s):abstr 3060.
69. Khan SA, Callahan M, Postow MA, et al. Ipilimumab in the treatment of uveal melanoma: the Memorial Sloan-Kettering Cancer Center experience. ASCO Meeting Abstracts. 2012;30(suppl):abstr 8549.
70. Luke JJ, Callahan MK, Postow MA, et al. Clinical activity of ipilimumab for metastatic uveal melanoma: a retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial Sloan-Kettering Cancer Center, and University Hospital of Lausanne experience. Cancer. 2013;119:3687-95.
71. Maio M, Danielli R, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann Oncol. 2013;24:2911-5.
72. Piulats Rodriguez JM, Ochoa de Olza M, Codes M, et al. Phase II study evaluating ipilimumab as a single agent in the first-line treatment of adult patients (Pts) with metastatic uveal melanoma (MUM): The GEM-1 trial. J Clin Oncol. 2014;32(suppl):abstr 9033.
73. Zimmer L, Vaubel J, Mohr P, et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS One. 2015;10:e0118564.
74. Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109-17.
75. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-30.
76. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
77. Kottschade LA, McWilliams RR, Markovic S, et al. The use of pembrolizumab for the treatment of metastatic uveal melanoma. ASCO Meeting Abstracts. 2015;33:9010.
78. Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397-405.
79. Carvajal RD, Piperno-Neumann S, Kapiteijn E, et al. SUMIT: phase III, randomized, placebo-controlled, double-blind trial of selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma. Late-breaking Abstract and Oral Presentation at the Society for Melanoma Research Congress. November 21, 2015. San Francisco, CA. http://www.melanomacongress.com/docs/SMR_2015_Congress_Late_Breaking_Abstracts.pdf. Accessed December 10, 2015.
80. Sacco JJ, Nathan PD, Danson S, et al. Sunitinib versus dacarbazine as first-line treatment in patients with metastatic uveal melanoma. J Clin Oncol. 2013;31(suppl):abstr 9031.
81. Abdel-Rahman MH, Boru G, Massengill J, et al. MET oncogene inhibition as a potential target of therapy for uveal melanomas. Invest Ophthalmol Vis Sci. 2010;51:3333-9.
82. Surriga O, Rajasekhar VK, Ambrosini G, et al. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12:2817-26.
83. Daud A, Kluger HM, Edelman G, et al. Activity of cabozantinib in metastatic uveal melanoma: updated results from a phase II randomized discontinuation trial (RDT). J Clin Oncol. 2013;31(suppl):abstr 9094.
84. Agarwala SS, Eggermont AM, O’Day S, Zager JS. Metastatic melanoma to the liver: a contemporary and comprehensive review of surgical, systemic, and regional therapeutic options. Cancer. 2014;120:781-9.
85. Rule W, Timmerman R, Tong L, et al. Phase I dose-escalation study of stereotactic body radiotherapy in patients with hepatic metastases. Ann Surg Oncol. 2011;18:1081-7.
86. Sato T. Locoregional management of hepatic metastasis from primary uveal melanoma. Semin Oncol. 2010;37:127-38.
87. Klingenstein A, Haug AR, Zech CJ, Schaller UC. Radioembolization as locoregional therapy of hepatic metastases in uveal melanoma patients. Cardiovasc Intervent Radiol. 2013;36:158-65.
88. Leyvraz S, Piperno-Neumann S, Suciu S, et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): a multicentric randomized trial. Ann Oncol. 2014;25:742-6.
89. Pinqpank JF, Hughes M, Alexander HR, et al. 9304 ORAL percutaneous hepatic perfusion (PHP) vs. best alternative care (BAC) for patients (pts) with melanoma liver metastases-efficacy update of the phase 3 trial (NCT00324727). Eur J Cancer. 2011;47(suppl 1):S653.
90. Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1998;83:1664-78.
91. Yu GP, Hu DN, McCormick S, Finger PT. Conjunctival melanoma: Is it increasing in the United States? Am J Ophthalmol. 2003;135:800-6.
92. Triay E, Bergman L, Nilsson B, et al. Time trends in the incidence of conjunctival melanoma in Sweden. Br J Ophthalmol. 2009;93:1524-8.
93. Tuomaala S, Eskelin S, Tarkkanen A, Kivela T. Population-based assessment of clinical characteristics predicting outcome of conjunctival melanoma in whites. Invest Ophthalmol Vis Sci. 2002;43:3399-408.
94. Hu DN, Yu G, McCormick SA, Finger PT. Population-based incidence of conjunctival melanoma in various races and ethnic groups and comparison with other melanomas. Am J Ophthalmol. 2008;145:418-23.
95. Shields CL, Markowitz JS, Belinsky I, et al. Conjunctival melanoma: outcomes based on tumor origin in 382 consecutive cases. Ophthalmology. 2011;118:389-95. e1-2.
96. Griewank KG, Westekemper H, Murali R, et al. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clin Cancer Res. 2013;19:3143-52.
97. Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821-8.
98. Wallander ML, Layfield LJ, Emerson LL, et al. KIT mutations in ocular melanoma: frequency and anatomic distribution. Mod Pathol. 2011;24:1031-5.
99. Populo H, Soares P, Rocha AS, et al. Evaluation of the mTOR pathway in ocular (uvea and conjunctiva) melanoma. Melanoma Res. 2010;20:107-17.
100. Lake SL, Jmor F, Dopierala J, et al. Multiplex ligation-dependent probe amplification of conjunctival melanoma reveals common BRAF V600E gene mutation and gene copy number changes. Invest Ophthalmol Vis Sci. 2011;52:5598-604.
101. Paridaens AD, McCartney AC, Minassian DC, Hungerford JL. Orbital exenteration in 95 cases of primary conjunctival malignant melanoma. Br J Ophthalmol. 1994;78:520-8.
102. Norregaard JC, Gerner N, Jensen OA, Prause JU. Malignant melanoma of the conjunctiva: occurrence and survival following surgery and radiotherapy in a Danish population. Graefes Arch Clin Exp Ophthalmol. 1996;234:569-72.
103. Wong JR, Nanji AA, Galor A, Karp CL. Management of conjunctival malignant melanoma: a review and update. Expert Rev Ophthalmol. 2014;9:185-204.
104. Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol. 1997;115:808-15.
105. Esmaeli B, Wang X, Youssef A, Gershenwald JE. Patterns of regional and distant metastasis in patients with conjunctival melanoma: experience at a cancer center over four decades. Ophthalmology. 2001;108:2101-5.
106. Tuomaala S, Kivela T. Metastatic pattern and survival in disseminated conjunctival melanoma: implications for sentinel lymph node biopsy. Ophthalmology. 2004;111:816-21.
107. Shields CL, Shields JA, Gunduz K, et al. Conjunctival melanoma: risk factors for recurrence, exenteration, metastasis, and death in 150 consecutive patients. Arch Ophthalmol. 2000;118:1497-507.
108. Missotten GS, Keijser S, De Keizer RJ, De Wolff-Rouendaal D. Conjunctival melanoma in the Netherlands: a nationwide study. Invest Ophthalmol Vis Sci. 2005;46:75-82.
109. Savar A, Ross MI, Prieto VG, et al. Sentinel lymph node biopsy for ocular adnexal melanoma: experience in 30 patients. Ophthalmology. 2009;116:2217-23.
110. Cohen VM, Tsimpida M, Hungerford JL, et al. Prospective study of sentinel lymph node biopsy for conjunctival melanoma. Br J Ophthalmol. 2013;97:1525-9.
111. Finger PT, Czechonska G, Liarikos S. Topical mitomycin C chemotherapy for conjunctival melanoma and PAM with atypia. Br J Ophthalmol. 1998;82:476-9.
112. Kurli M, Finger PT. Topical mitomycin chemotherapy for conjunctival malignant melanoma and primary acquired melanosis with atypia: 12 years’ experience. Graefes Arch Clin Exp Ophthalmol. 2005;243:1108-14.
113. Demirci H, McCormick SA, Finger PT. Topical mitomycin chemotherapy for conjunctival malignant melanoma and primary acquired melanosis with atypia: clinical experience with histopathologic observations. Arch Ophthalmol. 2000;118:885-91.
114. Ditta LC, Shildkrot Y, Wilson MW. Outcomes in 15 patients with conjunctival melanoma treated with adjuvant topical mitomycin C: complications and recurrences. Ophthalmology. 2011;118:1754-9.
115. Russell HC, Chadha V, Lockington D, Kemp EG. Topical mitomycin C chemotherapy in the management of ocular surface neoplasia: a 10-year review of treatment outcomes and complications. Br J Ophthalmol. 2010;94:1316-21.
116. Khong JJ, Muecke J. Complications of mitomycin C therapy in 100 eyes with ocular surface neoplasia. Br J Ophthalmol. 2006;90:819-22.
117. Finger PT, Sedeek RW, Chin KJ. Topical interferon alfa in the treatment of conjunctival melanoma and primary acquired melanosis complex. Am J Ophthalmol. 2008;145:124-9.
118. Herold TR, Hintschich C. Interferon alpha for the treatment of melanocytic conjunctival lesions. Graefes Arch Clin Exp Ophthalmol. 2010;248:111-5.
119. Stannard C, Sauerwein W, Maree G, Lecuona K. Radiotherapy for ocular tumours. Eye (Lond). 2013;27:119-27.
120. Cohen VM, Papastefanou VP, Liu S, et al. The use of strontium-90 Beta radiotherapy as adjuvant treatment for conjunctival melanoma. J Oncol. 2013;2013:349162.
121. Karim R, Conway RM. Conservative resection and adjuvant plaque brachytherapy for early-stage conjunctival melanoma. Clin Experiment Ophthalmol. 2011;39:293-8.
122. Stannard CE, Sealy GR, Hering ER, et al. Malignant melanoma of the eyelid and palpebral conjunctiva treated with iodine-125 brachytherapy. Ophthalmology. 2000;107:951-8.
123. Walsh-Conway N, Conway RM. Plaque brachytherapy for the management of ocular surface malignancies with corneoscleral invasion. Clin Experiment Ophthalmol. 2009;37:577-83.
124. Damato B, Coupland SE. An audit of conjunctival melanoma treatment in Liverpool. Eye (Lond). 2009;23:801-9.
125. Krause L, Ritter C, Wachtlin J, et al. [Recurrence rate following adjuvant strontium-90 brachytherapy after excision of conjunctival melanoma]. Klin Monbl Augenheilkd. 2008;225:649-52.
126. Wuestemeyer H, Sauerwein W, Meller D, et al. Proton radiotherapy as an alternative to exenteration in the management of extended conjunctival melanoma. Graefes Arch Clin Exp Ophthalmol. 2006;244:438-46.
127. Hsu A, Frank SJ, Ballo MT, et al. Postoperative adjuvant external-beam radiation therapy for cancers of the eyelid and conjunctiva. Ophthal Plast Reconstr Surg. 2008;24:444-9.
128. Spendlove HE, Damato BE, Humphreys J, et al. BRAF mutations are detectable in conjunctival but not uveal melanomas. Melanoma Res. 2004;14:449-52.
129. Paridaens AD, Minassian DC, McCartney AC, Hungerford JL. Prognostic factors in primary malignant melanoma of the conjunctiva: a clinicopathological study of 256 cases. Br J Ophthalmol. 1994;78:252-9.
130. Anastassiou G, Heiligenhaus A, Bechrakis N, et al. Prognostic value of clinical and histopathological parameters in conjunctival melanomas: a retrospective study. Br J Ophthalmol. 2002;86:163-7.
131. Werschnik C, Lommatzsch PK. Long-term follow-up of patients with conjunctival melanoma. Am J Clin Oncol. 2002;25:248-55.
132. Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-34.
133. Ambrosini G, Musi E, Ho AL, et al. Inhibition of mutant GNAQ signaling in uveal melanoma induces AMPK-dependent autophagic cell death. Mol Cancer Ther. 2013;12:768-76.
134. Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251-5.















![A third of patients had a response [to lifileucel], and of the patients who have a response, half of them were alive at the 4-year follow-up.](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fcancernetwork%2F6b7c9a3270c71a70749ba86000cfc78a29d74309-2988x1702.png%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=30 "A third of patients had a response [to lifileucel], and of the patients who have a response, half of them were alive at the 4-year follow-up.")








