Early Detection of Cutaneous Lymphoma
Cutaneous lymphomas comprise a spectrum of diseases characterized by infiltration of the skin by malignant lymphocytes. The clinical manifestations of cutaneous lymphomas vary, and they can mimic benign dermatoses,
ABSTRACT: Cutaneous lymphomas comprise a spectrum of diseases characterized by infiltration of the skin by malignant lymphocytes. The clinical manifestations of cutaneous lymphomas vary, and they can mimic benign dermatoses, as well as nodal or visceral malignancies with cutaneous spread. Cutaneous lymphomas are divided into T-cell lymphomas and B-cell lymphomas. Cutaneous T-cell lymphomas include mycosis fungoides, Szary syndrome, lymphomatoid papulosis, CD30+ large cell lymphoma, and adult T-cell leukemia/lymphoma. The extent and severity of skin manifestations in cutaneous T-cell lymphomas are prognostic indicators of extracutaneous involvement. Primary cutaneous B-cell lymphomas comprise 10% to 25% of all primary cutaneous non-Hodgkin's lymphomas and are classified according to their cell of origin. Most cutaneous B-cell lymphomas have an indolent course and excellent prognosis when compared to their nodal counterparts. Many factors have been implicated in the etiology of cutaneous lymphomas, including chemical and drug exposures, as well as microbial agents, such as the Epstein-Barr virus (EBV), human T-lymphocyte virus-1 (HTLV-1), and Borrelia burgdorferi. Immunohistochemistry and lymphocyte-receptor gene rearrangement studies are useful in distinguishing malignant from benign conditions. [ONCOLOGY 12(10):1521-1530, 1998]
Introduction
Primary cutaneous lymphomas encompass a wide spectrum of both T- and B-cell lymphomas. They are the second most common group of extranodal non-Hodgkin’s lymphomas after primary gastrointestinal lymphomas.[1] Primary cutaneous lymphomas have characteristic clinical behavior and histologic features, and their prognosis differs from that of primary nodal lymphomas of the same histologic subtype. Each case of cutaneous lymphoma must be assessed to determine whether it is of primary cutaneous origin or secondary to nodal and/or systemic disease.
Both cutaneous T-cell and B-cell lymphomas are further subdivided into different clinical entities. Within the spectrum of cutaneous T-cell lymphomas, mycosis fungoides is the most common subtype, comprising 50% of cases. Other less common clinical variants are the mycosis fungoides d’emblee variant, Woringer-Kolopp disease, Szary syndrome, alopecia mucinosa, poikiloderma vasculare atrophicans, lymphomatoid papulosis, CD30+ large cell lymphoma, and human T-lymphocyte virus type 1 (HTLV-1)-associated adult T-cell leukemia/lymphoma.
Cutaneous B-cell lymphomas are less common than cutaneous T-cell lymphomas and account for less than 20% of all primary cutaneous lymphomas.[2] Unlike the cutaneous T-cell lymphomas, the different subtypes of cutaneous B-cell lymphoma may appear similar clinically, but they are distinguishable at the histopathologic and molecular level.
Epidemiology and Causation
Cutaneous T-cell lymphoma is observed worldwide and affects both genders. Approximately 1,000 incident cases of cutaneous T-cell lymphoma occur annually in the United States. African-Americans experience the highest incidence rates, followed by Caucasians and Asian-Americans. Both incidence and mortality are higher in men than in women.[3,4]
Environmental Factors: Mycosis fungoides may be causally linked to chemical and nuclear exposures. A recent case-control study was conducted to determine whether exposure to chemicals in the workplace is related to an excess incidence of hematopoietic and lymphoid neoplasms. A positive association was found between exposure to chemicals and death due to non-Hodgkin’s lymphoma, multiple myeloma, and lymphoid leukemia at < 65 years of age.[5]
On the other hand, Whittemore et al found no consistent relationship between occupational or chemical exposure and risk for mycosis fungoides.[6] Another study conducted in Scotland showed no increased risk of childhood leukemia and non-Hodgkin’s lymphoma in cohorts of patients living near a nuclear site, but this study did not specifically mention cutaneous lymphoma.[7]
Infectious Pathogens: Evidence implicating viral infections as possible etiologic or contributing factors has been sought since the isolation of HTLV-1, a type-C retrovirus, from the lymphocytes of a patient with cutaneous T-cell lymphoma[8]. However, the restricted epidemiologic patterns (southwestern Japan, the West Indies, and southeastern United States) of HTLV-1 do not match the patterns of mycosis fungoides/Szary syndrome. In addition, serologic tests for HTLV-1 are negative in the majority of patients with mycosis fungoides/Szary syndrome.
The Epstein-Barr virus (EBV) genome has been found in B-cell lymphoma cells in persons with congenital or acquired immunodeficiency.[9] Chronic infection with Borrelia burgdorferi has also been associated with low-grade B-cell lymphoma,[10,11] while some cases of gastric B-cell lymphoma have regressed after eradication of Helicobacter pylori.[12]
Immunosuppression: Second malignancies occur with greater frequency in patients with cutaneous T-cell lymphoma, perhaps due to the immunosuppressive effect of the disease itself.[13] On the other hand, immunosuppression may be a risk factor for cutaneous T-cell lymphoma. Human immunodefic-iency virus type 1 (HIV-1) infection is associated predominantly with aggres-sive B-cell lymphomas, but mycosis fungoides has now been reported among HIV-1-positive individuals.[14]
Clinical Manifestations
FIGURE 1A


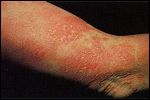
Patch-stage mycosis fungoides on the arm FIGURE 1B

Tumor-stage mycosis fungoides FIGURE 2

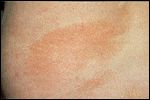
Parapsoriasis en Plaques
-Ill-defined, orange-brown patch of parapsoriasis on the flank
Mycosis fungoides is a low-grade lymphoma of mature CD4+ cells. It is characterized by three phases of evolution: patch, plaque, and tumor. Patients may have lesions from the three different phases simultaneously.
Initially, mycosis fungoides consists of single or multiple, erythematous, scaly patches, occurring mainly over the trunk and extremities (Figure 1A). These patches are often associated with severe pruritus. The patch stage may last from months to years before progressing to the plaque stage.
Plaques are elevated, well-demarcated lesions that are oval or circular in shape. They may regress spontaneously or coalesce to form larger plaques.
In the tumor stage, the neoplastic cells enter a vertical growth phase, leading to the clinical appearance of an expanding nodule (Figure 1B).Tumors of mycosis fungoides have a predilection for the face and skin folds (eg, axillae, groin, and inframammary areas in women). Central necrosis with ulceration can occur, resulting in secondary bacterial infections.
Parapsoriasis en plaques is clinically and histologically indistinguishable from early patch-stage mycosis fungoides. Cutaneous manifestations of the former include red-brown, atrophic, scaly patches with a wrinkled cigarette paper-like surface (Figure 2). The size of these plaques varies from 1 to > 6 cm in diameter, and they typically occur on the trunk. Some authors regard parapsoriasis as a latent prelymphoma with the potential to transform into mycosis fungoides, whereas others believe it is simply an early form of mycosis fungoides.[15]
The d’emblee variant of mycosis fungoides is a rare presentation in which tumors develop de novo without the long progression through patch and plaque stages. Affected patients tend to have an aggressive form of the disease and a poor prognosis.[16]
Woringer-Kolopp disease, also known as pagetoid reticulosis, presents as a single, erythematous, scaly plaque, usually on the extremities. Extracutaneous spread has not been reported.[17]
Szary syndrome, the leukemic variant of mycosis fungoides, constitutes approximately 5% of all newly reported cases of cutaneous T-cell lymphomas. It can arise de novo or as a progression from preexisting mycosis fungoides.
FIGURE 3

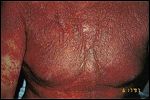
Szare Syndrome-
Erythroderma with infiltration of the skin by malignant lymphocytes
The malignant CD4+ cells circulate in the peripheral blood and populate the skin, creating diffuse erythroderma (Figure 3). Involvement of the face may lead to marked accentuation of facial folds, referred to as "leonine facies." There may be scaling and fissuring of the palms and soles, alopecia, ectro-pion, loss of nails, and ankle edema. The patient may complain of weight loss and malaise and may also experience fever and chills secondary to poor thermoregulation.
Examination of a peripheral blood smear shows hyperconvoluted lymphocytes with cerebriform nuclei, which, on immunophenotypic analysis, demonstrate a CD4+ CD7- phenotype. Investigators disagree as to how many circulating Szary cells are required to define the syndrome, but most agree that the consistent presence of more than 11% CD4+, CD7- lymphocytes, along with the appropriate clinical findings, can identify the disease.[18] Occasionally, Szary cells are encountered in patients with benign dermatoses, such as psoriasis and atopic dermatitis, and even in healthy individuals.[19]
Alopecia mucinosa, another form of T-cell infiltrative disease, consists of a primary form and a form that is associated with cutaneous T-cell lymphoma.[20] It manifests as grouped follicular papules or red, raised, boggy, nodular plaques located mainly on the head and neck. Hair loss over the scalp or eyebrows may be the presenting sign.
Poikiloderma vasculare atrophicans may be the first and only clinical manifestation of cutaneous T-cell lymphoma. It is characterized by reticulate hyperpigmentation and hypopigmentation, telangiectasia, and skin atrophy. Lesions vary in size and are distributed symmetrically, mostly affectingthe breasts, buttocks, and the major flexures.
Lymphomatoid papulosis, first described in 1968 by Macaulay, is a benign dermatosis, which, on pathology, mimics lymphoma.[21] Clinically, lymphomatoid papulosis is characterized by recurrent crops of red-brown papules occurring mainly on the trunk and extremities. The individual lesions, which resemble insect bites, develop necrotic centers and involute spontaneously, leaving behind hypopigmented or hyperpigmented scars. One crop can last 3 to 6 weeks, while the course of the disease may last for decades. The average age of onset is 40 years.
A subset of patients with lymphomatoid papulosis develop lymphoma (Hodgkin’s lymphoma, mycosis fungoides, or CD30+ large cell lymphoma).[22] These lymphomas can precede, follow, or occur concurrently with lymphomatoid papulosis. According to Cabanillas et al, the cumulative risk of transformation after 15 years is 80%.[23] Therefore, all patients with lymphomatoid papulosis require careful long-term follow-up and should be informed about clues that may indicate malignancy, such as persistent or enlarging nodules, lymphadenopathy, and the presence of B symptoms.[24]
FIGURE 4

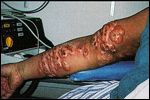
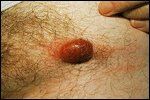
Primary Cutaneous CD30+ Large Cell Lymphoma-
Ulcerated nodules of primary cutaneous CD30+ large cell lymphoma localized to one arm
Cutaneous CD30+ lymphoproliferative disorders represent a recently recognized group of cutaneous lymphomas. They are divided into three groups: primary cutaneous CD30+ large cell lymphoma, secondary cutaneous CD30+ large cell lymphoma, and primary nodal CD30+ large cell lymphoma.[25] Primary cutaneous CD30+ large cell lymphoma generally occurs in adults and presents as solitary or multiple tumors localized to one site (Figure 4). The secondary cutaneous form develops in patients with a preexisting cutaneous lymphoma (mycosis fungoides or lymphomatoid papulosis) or can represent a cutaneous localization of a primary nodal CD30+ large cell lymphoma.[26]
Adult T-cell leukemia/lymphoma is sometimes included in the category of mycosis fungoides/Szary syndrome because the two share many features. First reported in Japan in 1977, adult T-cell leukemia/lymphoma cases are clustered in southwestern Japan, the West Indies, and southeastern United States.[27] Human T-lymphocyte virus-1 was isolated from patients with adult T-cell leukemia/lymphoma and was identified as the causative agent. Transmission occurs vertically from mother to child through breast-feeding and horizontally through sexual contact and shared needles.[28]
Variation in clinical features has led to the subclassification of adult T-cell leukemia/lymphoma into several types: pre-adult T-cell leukemia/lymphoma, which is completely asymptomatic; smoldering adult T-cell leukemia/lymphoma; chronic adult T-cell leukemia lymphoma; and acute adult T-cell leukemia/lymphoma, which is the most common form of presentation. Cutaneous lesions range from single to multiple papules and nodules, to large plaques, to generalized erythroderma.
Primary cutaneous B-cell lymphoma represents a distinct group of lymphoproliferative diseases that should be distinguished from non-Hodgkin’s B-cell lymphoma with secondary cutaneous involvement and from cutaneous pseudolymphomas. Primary cutaneous B-cell lymphoma is char-acterized by primary cutaneous presentation, the absence of extracutaneous disease, and the presence of light-chain monoclonal restriction.
FIGURE 5

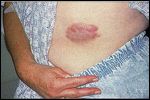
Primary cutaneous B-cell lymphoma of the abdomen
Primary cutaneous follicle center cell lymphoma accounts for 40% of primary cutaneous B-cell lymphomas.[29] Primary cutaneous follicle center cell lymphoma is characterized by plum-colored, firm, indurated nodules that have a smooth surface and lack scales (Figure 5). They may be single or multiple, widespread or aggregated, and most commonly occur on the back, head,and neck.
Large B-cell lymphoma of the leg has been recently described in elderly patients (> 70 years).[30] It presents with tumors on one or both legs and is associated with an intermediate prognosis.
Plasmacytomas are characterized by extramedullary proliferation of monoclonal plasma cells without an underlying multiple myeloma. The lesions are single or multiple, red nodules with no site predilection.[31] Immunocytomas, on the other hand, occur more commonly on the extremities.[32]
Intravascular cutaneous B-cell lymphoma can affect the skin, as well as the central nervous system. On the skin, it presents with violaceous, indurated plaques, usually on the lower extremities or the trunk.[33]
Extracutaneous Manifestations
Mycosis Fungoides/Szary Syndrome: In patients with mycosis fungoides/Szary syndrome, detection of extracutaneous disease is uncommon except in the later stages. Lymphade-nopathy is present in 47% of all patients (more often in those with tumor-stage mycosis fungoides) and in 80% to 90% of erythrodermic patients.[34]
TABLE 1


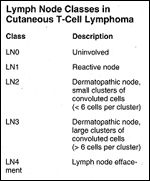
Lymph Node Classes in Cutaneous T-Cell Lymphoma
Sausville et al have developed a lymph node histopathologic classification system for cutaneous T-cell lymphoma, in which nodes are designated as LN0 to LN4, based on assessment of nodal architecture and extent of replacement by neoplastic lymphocytes (Table 1).[35] Lymph nodes showing preserved nodal architecture and scattered atypical cells (LN1) or small clusters of atypical lymphoid cells with convoluted nuclei (LN2) carry the most favorable prognosis. Nodes in which the nodal architecture is effaced by tumor cells (LN4) are associated with the least favorable clinical outcome.
Furthermore, detection of T-cell receptor gene rearrangement in lymph nodes has been shown to be of prognostic value. Patients with dermatopathic LN2/LN3 nodes in which the T-cell receptor gene rearrangement could not be detected had improved survival compared to those in whom this gene rearrangement was detectable at the level of Southern blotting.[36]
Pulmonary involvement is present in 40% to 60% of patients with mycosis fungoides/Szary syndrome at autopsy.[37] Pleural effusion with atypical T-cells has also been reported.
Bone involvement is seen in 30% to 40% of patients at autopsy, whereas bone marrow involvement by malignant T-cells is uncommon and increases with the stage of the disease.[38] Microscopic infiltration of the spleen and liver has been demonstrated in patients without clinically evident hepatosplenomegaly.[39]
Primary Cutaneous CD30+ Large Cell Lymphoma: A minority of patients with primary cutaneous CD30+ large cell lymphoma develop peripheral lymphadenopathy. Although there are reports of spontaneous regression of tumors, relapses after therapy are frequent. Nevertheless, most patients have a favorable prognosis, with a 4-year survival of 90%.[40]
Secondary large cell lymphoma arising in lesions of mycosis fungoides is associated with a less favorable prognosis. However, in a recent study of CD30+ large cell lymphoma that developed in patients with lymphomatoid papulosis, only those in whom extracutaneous CD30+ large cell lymphoma developed had a poor prognosis.[41]
Adult T-Cell Leukemia Lymphoma: Both the chronic and acute forms of adult T-cell leukemia/lymphoma are characterized by the following features: large numbers of circulating atypical CD4+ cells that overexpress the interleukin-2 (IL-2) receptor, lymphadenopathy, and hepatosplenomegaly. Hyper- calcemia, with or without lytic bone lesions, is found in approximately half of patients with acute adult T-cell leukemia/lymphoma. Many patients succumb to opportunistic infections, such as Pneumocystis carinii and cytomegalovirus pneumonia, cryptococcal meningitis, disseminated fungal infections, bacterial lung abscesses, and bacterial sepsis.[42]
Cutaneous B-Cell Lymphoma: A thorough work-up of patients with cutaneous B-cell lymphoma will reveal extracutaneous involvement in about 25% of patients.[43] Extracutaneous spread involves bone marrow, peripheral blood, liver, lungs, and other organs. Unlike cutaneous T-cell lymphoma, enlargement of peripheral lymph nodes in patients with cutaneous B-cell lymphoma usually indicates neoplastic infiltration as opposed to dermatopathic lymphadenopathy.[44]
Histopathology
FIGURE 6

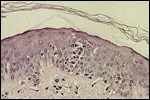
Pautrier's microabscess in a patient with Szary syndrome
Mycosis Fungoides: The histopathologic diagnosis of mycosis fungoides is based on the presence of the following features[45,46]: epidermotropism; large hyperchromatic lymphocytes within the epidermis and hair follicle; and Pautrier’s microabscesses, clusters of atypical lymphocytes in the epidermis (Figure 6). In early patch disease, the histopathologic picture may be nonspecific due to the scant lymphocytic infiltrate in the dermis.
FIGURE 7

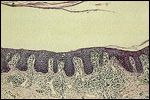
Band-like lymphocytic infiltrate along the dermoepidermal junction in a patient with mycosis fungoides
In the indurated plaque stage, the histology is more diagnostic, with a dermal infiltrate spanning the dermoepidermal junction in a band-like pattern (Figure 7). A high percentage of mycosis cells with hyperchromatic and irregularly shaped nuclei can be found in the epidermis and dermis.
In the tumor stage, the dermal infiltrate is heavy and often penetrates into the subcutaneous layer, compressing and destroying the overlying epidermis and resulting in superficial ulceration. Both the Szary syndrome and HTLV-1 disease have histopathologic features that are indistinguishable from patch/plaque mycosis fungoides.
FIGURE 8

CD30+ lymphocytes present in the infiltrate of lymphomatoid papulosis
Lymphomatoid papulosis has a characteristic wedge-shaped lymphocytic infiltrate that shows striking cytologic atypia. The atypical lymphocytes are divided into two types: type A cells, which have large vesicular nuclei resembling Reed-Sternberg cells, and type B cells, which resemble the cerebriform lymphocytes seen in mycosis fungoides. Immunohistochemical studies reveal that the type A cells are CD4+ and CD30+ (Figure 8), whereas the infiltrating lymphocytes in mycosis fungoides do not express the CD30 antigen, except in cases undergoing transformation to CD30+ large cell lymphoma.
FIGURE 9


Diffuse cellular infiltrate permeating the dermis of a patient with primary cutaenous CD30+ large cell lymphoma
CD30+ large cell lymphoma is characterized by a nonepidermotropic lymphocytic infiltrate, consisting of large CD30+ cells (Figure 9). In some patients, it may be difficult to distinguish between large cell lymphoma and lymphomatoid papulosis, both clinically and histologically. A helpful clue is the presence of sheets of CD30+ cells within the dermis in large cell lymphoma, as opposed to the occasional CD30+-positive cells seen in most cases of lymphomatoid papulosis.[47]
Cutaneous B-cell lymphomas are characterized by a sharply demarcated, nodular, bottom-heavy B-cell lymphocytic infiltrate in the mid- or deep dermis. As the infiltrates progress to cover larger areas, the nodular pattern is replaced by a diffuse one extending into the subcutaneous tissue. Immunophenotyping of the infiltrate in cutaneous B-cell lymphoma reveals expression of the pan B-cell antigens CD19 and CD20.[48]
Differential Diagnosis
TABLE 2


Differential Diagnosis of Patches and Plaques
Patches and Plaques: The presentation of early patch- or plaque-stage mycosis fungoides/Szary syndrome may frequently be confused with a number of benign skin conditions (Table 2). The erythematous, scaly lesions of early-stage mycosis fungoides can be seen in parapsoriasis, dermatitis, and psoriasis. These conditions can be histologically similar, since the subtle changes found in biopsies of early mycosis fungoides are difficult to distinguish from inflammatory changes. To further complicate matters, foci of atypical lymphocytes resembling Pautrier microabscesses, previously thought to be pathognomonic for mycosis fungoides, have been reported in patients with cutaneous fungal infections and chronic dermatitis.[49]
Immunohistochemistry and T-cell receptor gene rearrangement studies may help distinguish benign conditions from cutaneous T-cell lymphoma. In mycosis fungoides, there is antigen loss of the pan T-cell marker CD7 on CD4-expressing infiltrating tumor cells,[50] whereas in peripheral T-cell lymphoma of the skin, CD7 is often coexpressed with CD4.
Although all cutaneous T-cell lymphomas demonstrate clonality of T-cell gene rearrangement, clonality has also been reported in clinically benign skin disorders, such as pityriasis lichenoides and hydantoin-induced pseudolymphomas.[51,52] Therefore, clinicopatho-logic correlation and close patient follow-up are of utmost importance. It may be necessary to repeat skin biopsies if the clinical suspicion of lymphoma persists.
TABLE 3



Differential Diagnosis of Skin Tumors
Nodules of cutaneous lymphoma (such as are seen in patients with mycosis fungoides, CD30+ large cell lymphoma, or cutaneous B-cell lymphoma) can be confused with many benign and malignant entities (Table 3). Pseudolymphoma is a benign lymphoproliferative disorder with a number clinical features that distinguish it from malignant lymphomas:
1. Pseudolymphomas often present as a single papule or nodule, with a propensity for the face, neck, mammary area, scrotum, and arms. Lymphomas are associated with multiple tumors, are frequently ulcerated, and show no predilection for particular anatomic sites.
2. With pseudolymphomas, there may be a history of a causative event, such as insect bites, scabies, vaccination, acupuncture, tattoos, or drug intake. The culprit drugs are the hydantoin derivatives, carbamazepine, allopurinol, amiloride (Midamor), clomipramine (Anafranil), and cyclosporine (Neoral, Sandimmune).[53]
3. Pseudolymphomas have a tendency to regress spontaneously or after discontinuation of the causative agent. Lymphomas, on the other hand, are usually progressive.
The histologic differentiation of pseudolymphoma from cutaneous T-cell and B-cell lymphomas can be difficult. Cytologic features, such as nuclear atypia, increased number of mitotic figures, and even atypical mitotic figures, have been found in both conditions. Rearrangement of the genes encoding for surface immunoglobulins or the T-cell receptor on Southern blot testing may not be diagnostic of malignancy. Similarly, the presence of polyclonal B- or T-lymphocytes does not rule out malignancy.
It has been shown that in early lesions of mycosis fungoides and cutaneous B-cell lymphoma, the clonal T- and B-cells are hidden among the larger number of polyclonal reactive cells. The use of the polymerase chain reaction (PCR) instead of the Southern blot technique has greatly increased the sensitivity for detecting monoclonal lymphocytes early in the course of the disease; however, PCR techniques are not yet widely available.[54]
Erythroderma: The differential diagnosis of erythroderma includes drug reactions, pityriasis rubra pilaris, psoriasis, seborrheic dermatitis, and atopic dermatitis, all of which are benign entities. Less common causes of erythroderma include sarcoidosis, subacute cutaneous lupus erythematosus, and hepatitis. To differentiate these forms of erythroderma from the Szary syndrome, the physician must consider the clinical findings together with histopathologic, immunopathologic, and molecular genetic studies.
Staging, Course, and Prognosis
TABLE 4



TNM Staging of Cutaenous T-Cell Lymphoma
In 1979, the Mycosis Fungoides Cooperative Group proposed a TNM system to stage cutaneous T-cell lymphoma.[55] This staging system is shown in Table 4.
The extent and type of skin involvement and presence of extracutaneous disease are the most important predictors of survival in patients with mycosis fungoides. Researchers at Stanford Medical Center followed 122 patients with stage Ia mycosis fungoides for a median of 5 years and found no alterations in life expectancy. Of the patients with limited skin involvement and no extracutaneous involvement, only 10% to 15% progressed to more advanced cutaneous disease. The risk factors that were associated with disease progression included lack of clinical response to initial therapy and older age (> 60 years).[56]
Patients with extensive skin involvement have a less favorable prognosis. Multivariate analysis demonstrated a strong correlation between the primary tumor (skin) stage and lymph node stage: Patients with limited plaques do not have effaced nodes. In contrast, most patients with generalized skin involvement have evidence of advanced nodal disease (LN3/LN4).[57]
In patients with erythroderma, the most pertinent prognostic factors are age at presentation (patients < 65 years old do better), overall clinical stage, and the presence of peripheral blood involvement. Most patients with mycosis fungoides/Szary syndrome die from viral, fungal, or bacterial infections secondary to immunosuppression. Septicemia is prevalent during the late stages, when necrotic cutaneous lesions, invasive procedures, or radiodermatitis compromise the skin integument.[58] Aggressive therapy of cutaneous T-cell lymphoma may also enhance this immunosuppression.
In patients with adult T-cell leukemia/lymphoma, prognosis depends on the particular subtype: The survival rate at 4 years is 100% for patients with smoldering adult T-cell leukemia/lymphoma, 62.8% for those with the chronic form, and 5% for those with the acute form.[59]
Tashiro et al recognized that opportunistic infections occurred in HTLV-1 carriers and patients with smoldering adult T-cell leukemia/lymphoma who rapidly progressed to the acute form of the disease. They concluded that opportunistic infections may be predictive of the evolution of acute adult T-cell leukemia/lymphoma from the smoldering type.[60]
Primary cutaneous follicle center cell lymphoma and immunocytomas, which account for 90% of all primary cutaneous B-cell lymphomas, both share an excellent prognosis (5-year survival rate, > 95%) with a slight tendency toward extracutaneous spread.[61] Although histologically similar to primary cutaneous follicle center cell lymphoma, large B-cell lymphomas of the legs have a less favorable prognosis (5-year survival rate, 58%).[62] In addition, large B-cell lymphomas of the legs strongly express the bcl-2 protein, which is not found in primary cutaneous follicle center cell lymphoma, and this may be related to their less favorable outcome.
Work-up
After a diagnostic skin biopsy, a thorough physical examination and examination of peripheral blood should be performed in all patients with a cutaneous lymphoma. In patients with cutaneous B-cell lymphoma or CD30+ large cell lymphoma, computed tomographic (CT) scans should be done to rule out systemic adenopathy.
Patients with mycosis fungoides who have plaque disease and no adenopathy or involvement of the peripheral blood (as ascertained by immunophenotypic analysis) do not need further tests., as < 5% of these patients can be expected to have disease in visceral sites. Mycosis fungoides patients with adenopathy, circulating neoplastic lymphocytes, or T3 or T4 skin disease should undergo further evaluation for visceral involvement, including CT scans and biopsy of a palpable lymph node.
Biopsies should be submitted for light microscopy, flow cytometry, immunophenotyping, and T-cell receptor gene rearrangement or immunoglobulin light-chain gene rearrangement studies, if possible. Further evaluation of specific organ systems is indicated when involvement is suggested by history, examination, or routine tests.
In summary, the cutaneous lymphomas comprise a spectrum of both T- and B-cell lymphomas with varying histopathologic and clinical features. A careful history and use of immuno-histochemical and molecular diagnostic studies is crucial to accurately distinguish these disorders from benign dermatoses.
References:
1. Isaacson PG, Norton AJ: Cutaneous lymphoma, in Extranodal Lymphomas, p 172. London, Churchill Livingstone, 1994.
2. Willemze R, Kerl H, Sterry W, et al: EORTC classification for the primary cutaneous lymphomas: A proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood 90:354-371, 1997.
3. Weinstock MA, Gardstein B: Twenty year trends in the reported incidence of mycosis fungoides and its prognosis. J Invest Dermatol 108:544, 1997.
4. Weinstock MA, Horm JW: Mycosis fungoides in the United States: Increasing incidence and descriptive epidemiology. JAMA 260:42-46, 1988.
5. Massoudi BL, Talbott EO, Day RD, et al: A case-control study of hematopoietic and lymphoid neoplasms: The role of work in the chemical industry. Am J Ind Med 31:21-27, 1997.
6. Whittemore AS, Holly EA, Lee IM, et al: Mycosis fungoides in relation to environmental exposures and immune response: A case-control study. J Natl Cancer Inst 81:1560-1567, 1989.
7. Sharp L, Black RJ, Harkness EF, et al: Incidence of childhood leukemia and non-Hodgkin’s lymphoma in the vicinity of nuclear sites in Scotland. Occup Environ Med 53:823-831, 1996.
8. Poiesz BJ, Ruscetti FW, Gazdar AF, et al: Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T cell lymphoma. Proc Natl Acad Sci USA 77:7415-7419, 1980.
9. Levine AM: Acquired immunodeficiency syndrome-related lymphoma. Blood 80:8-20, 1992.
10. Kutting B, Bonsmann G, Metze D, et al: Borrelia burgdorferi-associated primary cutaneous B cell lymphoma: Complete clearing of skin lesions after antibiotic pulse therapy or intralesional injection of interferon alfa-2a. J Am Acad Dermatol 36:311-314, 1997.
11. Garbe C, Stein H, Gollnick H, et al: Cutaneous B-cell lymphoma in chronic Borrelia burgdorferi infection: Report of two cases and a review of the literature. Hautarzt 39:717-726, 1988.
12. Bayerdorffer E, Neubauer A, Rudolph B, et al: Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 345:1591-1594, 1995.
13. Olsen EA, Delzell E, Jegasothy B: Second malignancies in cutaneous T cell lymphoma. J Am Acad Dermatol 10:197-204, 1984.
14. Burns MK, Cooper KD: Cutaneous T-cell lymphoma associated with HIV infection. J Am Acad Dermatol 29:394-399, 1993.
15. Kikuchi A, Naka W, Harada T, et al: Parapsoriasis en plaques: Its potential for progression to malignant lymphoma. J Am Acad Dermatol 29:419-422, 1993.
16. Carlotti A, Dallot A, Laroche L, et al: Cutaneous lymphomas of the T cell type presenting primary tumors. Curr Probl Dermatol 19:157-160, 1990.
17. Burns MK, Chan LS, Cooper KD: Woringer-Kolopp disease (localized pagetoid reticulosis) or unilesional mycosis fungoides? An analysis of eight cases with benign disease. Arch Dermatol 131:325-329, 1995.
18. Bogen SA, Pelley D, Charif M, et al: Immunophenotypic identification of Sezary cells in peripheral blood. Am J Clin Pathol 106:739-748, 1996.
19. Stolz W, Schmoeckel C, Burg G, et al: Circulating Sezary cells in the diagnosis of Sezary syndrome (quantitative and morphometric analyses). J Invest Dermatol 81:314-319, 1983.
20. Gibson LE, Muller SA, Leiferman KM, et al: Follicular mucinosis: Clinical and histopathologic study. J Am Acad Dermatol 20:441-446, 1989.
21. Macaulay WL: Lymphomatoid papulosis: A continuing self-healing eruption, clinically benign-histologically malignant. Arch Dermatol 97:23-30, 1968.
22. McCarty MJ, Vukelja SJ, Sausville EA: Lymphomatoid papulosis associated with Ki-1-positive anaplastic large cell lymphoma: A report of two cases and a review of the literature. Cancer 74:3051-3058, 1994.
23. Cabanillas F, Armitage J, Pugh WC, et al: Lymphomatoid papulosis: A T-cell dyscrasia with a propensity to transform into malignant lymphoma. Ann Intern Med 122:210-217, 1995.
24. Demierre MF, Goldberg LJ, Kadin ME, et al: Is it lymphoma or lymphomatoid papulosis? J Am Acad Dermatol 36:765-772, 1997.
25. Willemze R, Beljaards RC: Spectrum of primary cutaneous CD30+ (Ki-1)-positive lymphoproliferative disorders. J Am Acad Dermatol 28:973-980, 1993.
26. Kaudewitz P, Stein H, Dallenbach F, et al: Primary and secondary cutaneous Ki-1+ (CD30+) anaplastic large cell lymphomas: Morphologic, immunohistologic, and clinical characteristics. Am J Pathol 135:359-367, 1989.
27. Cruickshank JK, Corbin DO, Bucher B, et al: HTLV-1 and neurological disease. Baillieres Clin Neurol 1:61-81, 1992.
28. Hollsberg P, Hafler DA: Seminars in medicine of the Beth Israel Hospital, Boston: Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med 328:1173-1182, 1993.
29. Willemze R: New concepts in the classification of cutaneous lymphomas. Arch Dermatol 131:1077-1080, 1995.
30. Vermeer MH, Geelen FA, van Haselen CW, et al: Primary cutaneous large B-cell lymphoma of the legs: A distinct type of cutaneous lymphoma with an intermediate prognosis: Dutch Cutaneous Lymphoma Working Group. Arch Dermatol 132:1304-1308,1996.
31. Chang YT, Wong CK: Primary cutaneous plasmacytomas. Clin Exp Dermatol 19:177-180, 1994.
32. LeBoit PE, McNutt NS, Reed JA, et al: Primary cutaneous immunocytoma: A B-cell lymphoma that can easily be mistaken for cutaneous lymphoid hyperplasia. Am J Surg Pathol 18:969-978, 1994.
33. Wick MR, Mills SE: Intravascular lymphomatosis: Clinicopathologic features and differential diagnosis. Semin Diagn Pathol 8:91-101, 1991.
34. Carney DN, Bunn PA Jr: Manifestations of cutaneous T-cell lymphoma. J Dermatol Surg Oncol 6:369-377, 1980.
35. Sausville EA, Worsham GF, Matthews MJ, et al: Histologic assessment of lymph nodes in mycosis fungoides/Sezary syndrome (cutaneous T-cell lymphoma): Clinical correlations and prognostic import of a new classification system. Hum Pathol 16:1098-1109, 1985.
36. Lynch JW Jr, Linoilla I, Sausville EA, et al: Prognostic implications of evaluation for lymph node involvement by T-cell antigen receptor gene rearrangement in mycosis fungoides. Blood 79:3293-3299, 1992.
37. Stokar LM, Vonderheid EC, Abell E, et al: Clinical manifestations of intrathoracic cutaneous T-cell lymphoma. Cancer 56:2694-2702, 1985.
38. O’Reilly GV, Clark TM, Crum CP: Skeletal involvement in mycosis fungoides. Am J Roentgenol 129:741-743, 1977.
39. Griem ML, Moran EM, Ferguson DJ, et al: Staging procedures in mycosis fungoides. Br J Cancer 31(suppl):362-367, 1975.
40. Paulli M, Bert E, Rosso R, et al: CD30+/Ki-1-positive lymphoproliferative disorders of the skin: Clinicopathologic correlation and statistical analysis of 86 cases: A multicentric study from the European Organization for Research and Treatment of Cancer Cutaneous Lymphoma Project Group. J Clin Oncol 13:1343-1354, 1996.
41. Beljaards RC, Willemze R: The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br J Dermatol 126:596-602, 1992.
42. Rhew DC, Gaultier CR, Daar ES, et al: Infections in patients with chronic adult T-cell leukemia lymphoma: Case report and review. Clin Infect Dis 21:1014-1018, 1995.
43. Burg G, Kaudewitz P: Where are we today in the diagnosis of cutaneous lymphoma? Curr Probl Dermatol 19:90-104, 1990.
44. Santucci M, Pimpinelli N, Arganini L: Primary cutaneous B-cell lymphoma: A unique type of low-grade lymphoma. Cancer 67:2311-2326, 1991.
45. Chu A, Berger CL, Kung P, et al: In situ identification of Langerhans cells in the dermal infiltrate of cutaneous T cell lymphoma. J Am Acad Dermatol 6:350-354, 1982.
46. Sanchez JL, Ackerman AB: The patch stage of mycosis fungoides: Criteria for histologic diagnosis. Am J Dermatopathol 1:5-26, 1979.
47. Smoller BR, Glusac EJ: Histologic mimics of cutaneous lymphoma. Pathol Annu 30:123-141, 1995.
48. Burg G, Kaudewitz P, Klepzig K: Cutaneous B-cell lymphoma. Dermatol Clin 3:689-704.
49. Ackerman AB, Breza TS, Capland L: Spongiotic stimulants of mycosis fungoides. Arch Dermatol 109:218-220, 1974.
50. Wallace ML, Smoller BR. Immunohistochemistry in diagnostic dermatopathology. J Am Acad Dermatol 34:163-183, 1996.
51. Zelickson BD, Peters MS, Muller SA, et al: T-cell receptor gene rearrangement analysis: cutaneous T cell lymphoma, peripheral T-cell lymphoma, and premalignant and benign cutaneous lymphoproliferative disorders. J Am Acad Dermatol 25:787-796, 1991.
52. D’Incan M, Souteyrand P, Bignon YJ, et al: Hydantoin-induced cutaneous pseudolymphoma with clinical, pathologic, and immunologic aspects of Sezary syndrome. Arch Dermatol 128:1371-1374,1992.
53. Callot V, Roujeau JC, Bagot M, et al: Drug-induced pseudolymphoma and hypersensitivity syndrome: Two different clinical entities. Arch Dermatol 132:1315-1321, 1996.
54. Crowson AN, Magro CM: Cutaneous pseudolymphoma: A review. J Clin Dermatol 3:43-55, 1995.
55. Bunn PA Jr, Lamberg SI: Report of the Committee on Staging and Classification of Cutaneous T-cell Lymphoma. Cancer Treat Rep 63:725-729, 1979.
56. Kim YH, Jensen RA, Watanabe GL: Clinical stage IA (limited patch and plaque) mycosis fungoides: A long term outcome analysis. Arch Dermatol 132:1309-1313, 1996.
57. Kim YH, Bishop K, Varghese A: Prognostic factors in erythrodermic mycosis fungoides and the Sezary syndrome. Arch Dermatol 131:1003-1008, 1995.
58. Posner LE, Fossiek BE Jr, Eddy JL, et al: Septicemic complications of the cutaneous T cell lymphomas. Am J Med 71:210-216, 1981.
59. Shimoyama M: Diagnostic criteria and classification of clinical subtypes of adult T-cell leukemia lymphoma. Br J Haematol 79:428-437, 1991.
60. Tashiro T, Yamasaki T, Nagai H, et al: Immunological studies on opportunistic infections and the development of adult T-cell leukemia. Intern Med 31:1132-1136, 1992.
61. Pimpinelli N, Santucci M, Mori M, et al: Primary cutaneous B-cell lymphoma: A clinically homogeneous entity? J Am Acad Dermatol 37:1012-1016, 1997.
62. Kerl H, Cerroni L: The morphologic spectrum of cutaneous B-cell lymphomas. Arch Dermatol 132:1376-1377, 1996.














