Although pediatric acute lymphoblastic leukemia (ALL) has cure rates of over 90%, adult ALL remains a challenging disease to treat, with cure rates roughly half those seen in children. The inferior outcomes in adults can be attributed mainly to adverse genetic features, as well as the inability-particularly of older adults-to tolerate chemotherapy. Modest improvements have been seen in outcomes for adolescents and young adults; these can largely be attributed to the use of pediatric-type combination chemotherapy regimens in patients aged 50 years or younger. In patients with Philadelphia chromosome–positive ALL, once a very-high-risk group, outcomes have markedly improved as a result of the use of tyrosine kinase inhibitors in combination with chemotherapy. The persistence of minimal residual disease has emerged as the single most important prognostic factor for ALL and is increasingly being used to help make decisions regarding allogeneic hematopoietic stem cell transplantation or novel salvage therapies. Relapsed/refractory ALL has had a dismal prognosis. In recent years, novel immune-based therapies have been developed that have shown impressive results and that have the potential to improve the outcome of relapsed ALL. These include antibody-drug conjugates, the bispecific T-cell–engaging antibody blinatumomab, and chimeric antigen receptor–modified T cells.
Introduction
Although acute lymphoblastic leukemia (ALL) is most commonly a pediatric malignancy, nearly half of cases occur in adults.[1] While exceptional progress has been made in curing pediatric ALL, with cure rates exceeding 90% in recent reports,[2] outcomes for adult ALL remain considerably inferior. There is still no consensus regarding the best treatment approach for newly diagnosed adults, and this is particularly true for older patients. Here, we discuss the challenges and new advances in adult ALL, as well as our approach to the treatment of these patients.
Epidemiology
Over 6,500 cases of ALL are projected to be diagnosed in the United States in 2016, of which 40% will be in persons older than 18 years of age.[1,3] The majority of ALL cases are classified as precursor B-cell type, but the T-cell phenotype peaks in the fourth decade of life and represents up to 25% of adult ALL cases.[4] T-cell ALL is slightly more common in adults than in children, and it appears more common in black persons than in whites.[4,5] The incidence rate of ALL is higher in Hispanics, with an incidence rate ratio of 2 compared with whites in the 20- to 54-year age group.[1,6] This is likely due to the higher incidence of the Philadelphia chromosome (Ph)-like ALL subset in Hispanics, which in turn appears to be related to a higher prevalence of specific inherited GATA3 variants in this ethnic group.[7,8]
Why Are Outcomes in Adults With ALL Inferior to Outcomes in Children?
Adolescent and young adult (AYA) ALL has long-term survival rates that are roughly half those seen in pediatric ALL. Various leukemia-, patient-, and treatment-related factors could explain this disparity. Most of the disparity can be explained by the adverse genetic makeup of adult ALL; underutilization of pediatric-type chemotherapy regimens in adults; and comorbidities and treatment side effects, which result in the administration of lower cumulative doses of active agents. Another major issue is the fact that many adult patients with ALL are cared for outside of specialized centers, and given the rarity of adult ALL, many oncologists have limited experience with its management. Specifically, oncologists who care for adults have less familiarity with risk stratification, administration of complicated ALL regimens, and management of these regimens’ toxicities compared with pediatric oncologists. In addition, oncologists who care for adults tend to adhere less strictly to regimens in terms of maintaining cycle schedules and delivering drugs on time. In part this is because ALL therapy in adult patients can lead to significant disruption of work and family life for a prolonged period of time, which in turn can result in significant noncompliance with treatment and follow-up appointments (unlike with pediatric patients, whose parents usually ensure that they attend visits and receive treatment on time). The fact that most adults are treated outside of clinical trials also contributes to less stringent adherence to treatment schedules and regimens. Finally, adult patients also tend to have significant comorbidities (in some cases prior malignancies, with resultant chemotherapy exposure) that further compromise chemotherapy.
The Genetic Landscape of Adult ALL
The genetic profile of ALL is very heterogeneous and encompasses recurrent chromosomal aberrations as well as gene mutations. Cytogenetics at the time of diagnosis is the single most important prognostic factor for patients treated upfront with chemotherapy regimens alone, and it is routinely used to risk-stratify patients and to make decisions regarding the use of allogeneic hematopoietic stem cell transplantation (HSCT) as consolidation therapy.[9-11]
In contrast to pediatric ALL, ALL in adults is characterized by a higher frequency of high-risk cytogenetics and a lower incidence of favorable genetic abnormalities. The Philadelphia chromosome, which refers to the translocation t(9;22)(q34;q11), is the most common cytogenetic abnormality, observed in around 25% of adults; the incidence of Ph+ ALL peaks in the fourth decade and plateaus afterwards.[12] Ph+ ALL is considered a very-high-risk subset, associated with a low response rate and a brief duration of remission; however, its prognosis has considerably improved with the routine addition of tyrosine kinase inhibitors (TKIs) to chemotherapy.
Ph-like ALL is a newly recognized entity that has a gene expression profile similar to that of Ph+ ALL, but without t(9;22). The incidence of Ph-like ALL peaks in young adults and then decreases in subsequent years, while the incidence of Ph+ ALL increases in older adults.[13,14] Ph-like ALL carries a poor prognosis, much like Ph+ ALL, but more than 90% of patients have kinase-activating mutations, potentially making them amenable to TKI therapy.[13,15] Around half of patients with Ph-like ALL have a rearrangement of CRLF2, and of these, half carry a JAK2 mutation.[13,16,17] In adults, CRLF2 rearrangement was observed in 15% of high-risk B-cell ALL, and it was correlated with adverse outcomes.[18,19] Deletion of IKZF1 is observed at higher frequencies in Ph+ ALL and Ph-like ALL,[13] and it was correlated with inferior prognosis in both these populations; however, patients with IKZF1 mutations appear to benefit from early allogeneic HSCT.[20-22]
The MLL gene on chromosome 11q23 is rearranged in 7% of adults with ALL. This abnormality is associated with an inferior prognosis; patients usually present with a high white blood cell count, aberrant expression of myeloid markers, and a lack of CD10 expression.[9-11] Much as in therapy-related acute myeloid leukemia, the MLL gene rearrangement is a common genetic abnormality observed in patients in whom ALL develops following prior cytotoxic therapy (therapy-related ALL).[23-25] MLL gene rearrangements can be seen in up to 8% of adult patients with T-cell ALL, most commonly t(11;19).[26,27]
The Notch signaling pathway is critical for T-cell development. Almost two-thirds of patients with T-cell ALL harbor mutations in Notch signaling pathway genes, and another 15% have either deletions or mutations in FBXW7, a gene that impairs the degradation of activated Notch 1 in the nucleus.[28]
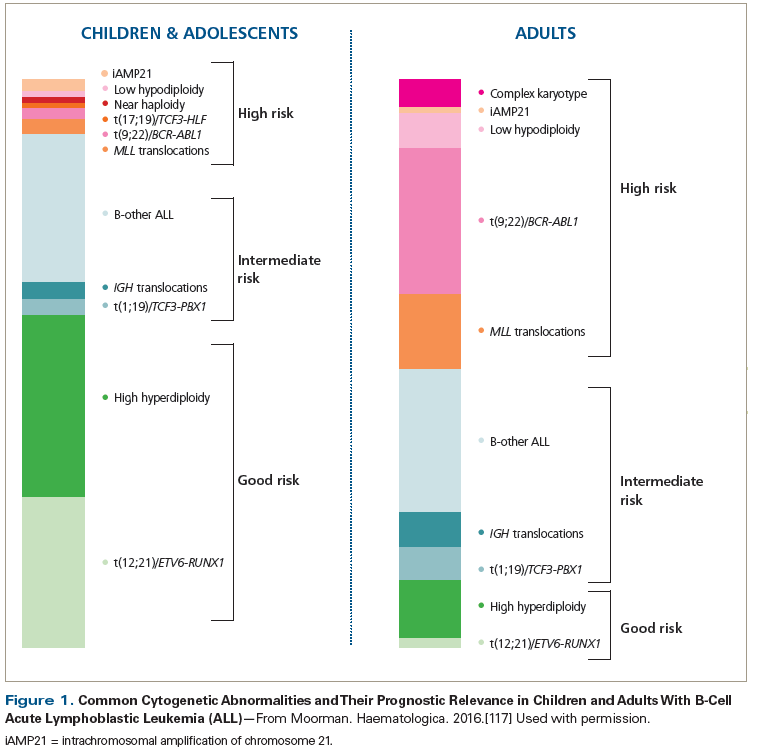
Interestingly, among patients with early thymic T-cell ALL (identified by its distinctive phenotype [CD1a(−), CD8(−), CD5(weak)] and expression of stem-cell or myeloid markers), FLT3 mutations are common and can be seen in up to one-third of cases; the majority of these are internal tandem duplication mutations of FLT3.[29] High hyperdiploidy (> 50 chromosomes) and t(12;21) (ETV6-RUNX1) are classified as favorable findings associated with better outcomes, but they are more frequently encountered in children than in adults.[10,12] Other cytogenetic abnormalities seen include complex karyotype, hypodiploidy, TCF3-PBX1 [t(1;19)], and IGH rearrangements. A listing of the more common cytogenetic abnormalities and their prognostic significance is provided in Figure 1 and discussed in references 9–12, 23–25, and 30 and 31.
Treatment of Newly Diagnosed Adult ALL
Curing ALL with chemotherapy requires a distinctive treatment approach that differs fundamentally from the principles of curative therapy for hematologic malignancies. Prolonged exposure to relatively low single doses of antimetabolites (resulting in high cumulative doses of individual agents) is the hallmark of pediatric-type ALL regimens. The majority of the progress in adult ALL in recent years can be attributed to the application of pediatric-type regimens to adult ALL patients, as a result of which 5-year survival rates of 50% to 60% have been achieved. Most of these regimens are modeled on the Berlin-Frankfurt-Münster regimen backbone. The principles of such regimens are listed in the Table. Adult-type regimens, on the other hand, are usually modeled after the hyper-CVAD regimen (see the following section for specifics of this regimen), which uses alternating cycles of active agents in higher individual doses that result in more myelosuppression. Induction cycles usually carry the highest risk of treatment-related complications. Morphologic complete remission (CR; defined as < 5% bone marrow blasts) is typically achieved readily, irrespective of the regimen used. However, relapse remains common in adults and is the leading cause of mortality.
Pediatric-type regimens include 1 to 2 cycles of induction (with the initial induction comprised of an anthracycline, prednisone, vincristine, and asparaginase). For patients who achieve morphologic remission, consolidation with either allogeneic HSCT or repeated cycles of combination chemotherapy is pursued. For those who are selected for chemotherapy consolidation, repeated combination chemotherapy cycles are administered over a period of 6 to 8 months. Most consolidation cycles in pediatric-derived regimens are administered in the outpatient setting, with the exception of high-dose methotrexate, which requires hospitalization for intensive hydration. The duration of neutropenia is generally brief with contemporary regimens, and the use of growth factors is encouraged.[32,33] Early and intensive intrathecal prophylaxis is a critical component of pediatric-type ALL regimens. Consolidation is followed by a prolonged period (2 to 3 years) of low-dose maintenance therapy, the goal of which is to eradicate small numbers of residual leukemia cells. The main components of maintenance therapy are daily 6-mercaptopurine and weekly oral methotrexate, combined frequently with monthly corticosteroid pulses and vincristine during the first year. While receiving maintenance therapy, patients usually return to their original lifestyle, with limited restrictions. The critical importance of adherence to 6-mercaptopurine maintenance therapy is well established and was highlighted in a recent study of pediatric ALL.[34]
Treatment of Young and Middle-Aged Adults With ALL
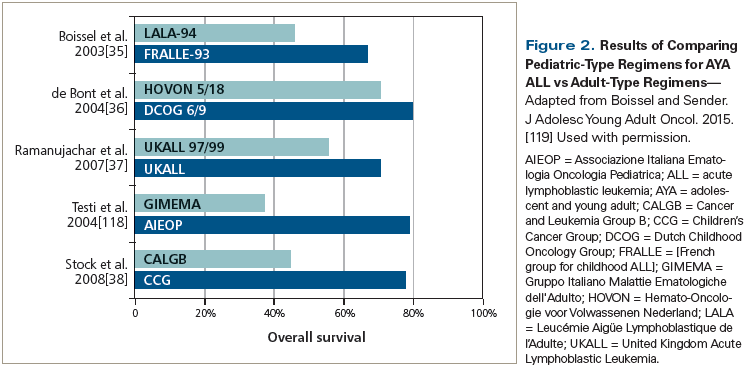
AYA patients with ALL, defined by the National Cancer Institute as adults under 40 years of age, comprise a distinct subgroup with regard to upfront therapy, given the ability of many in this age group to tolerate pediatric-type regimens. In fact, most of the recent success in therapy for newly diagnosed adult ALL can be attributed to the use of such regimens in this age group. Retrospective comparative data have consistently revealed better outcomes with pediatric-type regimens vs adult-type regimens, at least in adolescents and younger adults,[35-38] and emerging data indicate the ability of adults older than 40 years of age to tolerate modified pediatric regimens.[39-41] Figure 2 highlights the survival difference between pediatric-type and adult-type regimens in AYA patients with ALL. The 2-year event-free survival (EFS) and overall survival (OS) rates of 296 evaluable young adult patients with ALL enrolled in the US Intergroup C10403 trial were as high as 66% and 78%, respectively.[41] When pediatric-inspired regimens are used in adults, allogeneic HSCT in first complete remission (CR1) cannot be universally recommended for consolidation. A recent analysis indicated superior survival with a pediatric-type ALL regimen compared with matched registry data for allogeneic HSCT in younger adults (aged 18 to 50 years) with ALL in CR1.[42]
The practical definition of young adults, with respect to ability to tolerate pediatric regimens, remains to be determined and has varied among studies. The Programa Español de Tratamientos en HematologÃa (PETHEMA) group enrolled patients up to age 30[43]; the Intergroup study and the Hemato-Oncologie voor Volwassenen Nederland (HOVON) trial enrolled patients up to age 40 years[41,44]; while the Dana-Farber Cancer Institute study treated patients up to 50 years of age.[40] On the other hand, the Group for Research in Adult ALL (GRAALL) 2003 study and other studies have safely extended pediatric-type ALL treatment up to age 60.[39,45,46] Thus, the administration of a modified pediatric regimen appears feasible in adults at least up to age 60 if they have no significant comorbidities or organ dysfunction, with the ultimate goal of curing these patients without the need for allogeneic HSCT.
Methotrexate is a critical component of ALL regimens. Data support escalating methotrexate doses in children and young adults with ALL.[47,48] Similar benefit from high-dose methotrexate is expected in adult ALL as well,[49] and it is generally a well-tolerated drug even in older adults, if used judiciously with particular attention to renal function.
Hyper-CVAD is one of the most widely used regimens in adults with ALL, especially outside of the academic setting. This regimen consists of alternating cycles of myelotoxic drugs for 8 rounds (part A: cyclophosphamide, doxorubicin, vincristine, and dexamethasone; and part B: high-dose methotrexate and high-dose cytarabine), which is followed by maintenance. Hyper-CVAD does not include asparaginase and it requires frequent hospitalizations. Although a formal direct comparison has not been carried out, data indicate superior outcomes with pediatric-inspired regimens compared with hyper-CVAD in adult ALL.[50-52] Additionally, the repeated episodes of severe and prolonged neutropenia seen with hyper-CVAD predispose patients to serious bacterial and fungal infections. A recent retrospective study highlighted the high risk of relapse in T-cell ALL when patients are treated with the hyper-CVAD regimen, especially when no consolidation with allogeneic HSCT follows.[53]
Expression of CD20 is seen in about 30% of cases of B-cell ALL.[54] The addition of the anti-CD20 monoclonal antibody rituximab improved survival of younger adults who were treated with the hyper-CVAD regimen in a nonrandomized study.[55] In the randomized GRAALL-R 2005 study, rituximab was associated with better EFS when combined with a pediatric-inspired regimen, and resulted in modest OS prolongation in patients who did not undergo allogeneic HSCT in CR1.[56]
The Use of Asparaginase in Adult ALL
Asparaginase is a bacterial enzyme that converts asparagine into aspartic acid and ammonia. ALL cells are unable to synthesize asparagine and therefore depend on plasma asparagine for protein synthesis. Consequently, asparagine depletion leads to apoptosis of ALL cells. Among asparaginase preparations, polyethylene glycol–conjugated asparaginase (PEG-asparaginase) has the longest half-life, and it has replaced the short-acting Escherichia coli asparaginase.
Asparaginase has long been perceived as a toxic drug in adults; as a result, many adult regimens have not included it or have used it in inadequate doses. Although there is an increased incidence of certain asparaginase-related toxicities with age, these are manageable for the most part and do not justify omitting this key drug from contemporary ALL regimens.[57] Hepatotoxicity is common with the use of asparaginase in adults, and high-grade hyperbilirubinemia and transaminitis were observed in a quarter and half of treated patients, respectively. The risk of hyperbilirubinemia appears higher in early cycles and increases with age and body mass index.[58] Although hyperbilirubinemia can delay administration of subsequent cycles of therapy, this complication is always reversible and should not preclude administration of additional asparaginase doses, since the risk of recurrence is low in subsequent cycles. Clinical pancreatitis was observed in 13% of adults treated with asparaginase.[58] Due to the substantial risk of recurrence and serious complications, it is contraindicated to re-administer asparaginase to patients who have experienced clinical pancreatitis. High-grade hypertriglyceridemia is very common with asparaginase (51%) beyond the first cycle, but this is usually reversible and is not correlated with the occurrence of pancreatitis.[58]
Venous thrombosis is more common than bleeding during asparaginase therapy and occurs in 12% of patients. Thrombosis risk increases with age and usually occurs early in the treatment course.[57,58] The risk of thrombosis recurrence is negligible in patients who are maintained on anticoagulation during subsequent doses of asparaginase[58]; however, the use of prophylactic anticoagulation or coagulation factor/antithrombin III concentrate replacement remains controversial.[59,60] Despite hypofibrinogenemia being very common during asparaginase use (fibrinogen level < 100 mg/dL in 48%), bleeding risk is low and does not justify routine cryoprecipitate replacement, due to the risk of inducing thrombosis.[58]
Hypersensitivity reactions to asparaginase involve the generation of antibodies that inactivate the drug. Hypersensitivity reaction to asparaginase is observed less frequently with the pegylated form and has been reported less often in adults.[57] However, the liberal use of corticosteroids in adults could be masking clinical manifestations of hypersensitivity. Erwinia asparaginase is now approved for patients who develop a hypersensitivity reaction to E coli asparaginase, but this drug has a short half-life and 6 doses need to be given on alternate days to equal 1 dose of PEG-asparaginase. Silent hypersensitivity can result in loss of drug activity and can only be detected by measurement of asparaginase activity. Obtaining these measurements is becoming common clinical practice; the target level on day 14 post PEG-asparaginase dose is > 0.1 IU/mL.
Treatment of Older Adults With ALL
ALL in older adults carries a dismal prognosis due to both disease-related factors and the inability of many of these patients to tolerate full-intensity therapy because of its toxicity.[61] Nonetheless, ALL is curable in a subset of older patients, and every effort should be made to attempt curative therapy whenever possible. This can be done through modification or elimination of certain agents in order to improve tolerability for this age group, routine use of TKIs in Ph+ ALL, early referral for reduced-intensity allogeneic HSCT, and introduction of novel therapies into the chemotherapy backbone.
TO PUT THAT INTO CONTEXT
[[{"type":"media","view_mode":"media_crop","fid":"55090","attributes":{"alt":"","class":"media-image","id":"media_crop_1253218895262","media_crop_h":"0","media_crop_image_style":"-1","media_crop_instance":"6893","media_crop_rotate":"0","media_crop_scale_h":"0","media_crop_scale_w":"0","media_crop_w":"0","media_crop_x":"0","media_crop_y":"0","style":"height: 144px; width: 144px;","title":"","typeof":"foaf:Image"}}]]
Robert J. Soiffer, MD
Dana-Farber Cancer Institute
Boston, MassachusettsWhat New Options Are Improving Outcomes for Adults With ALL?After decades of only modest progress in the treatment of acute lymphoblastic leukemia (ALL), particularly for adults, the past 5 to 10 years have seen an explosion of new therapeutic approaches that have changed the landscape of treatment. One of these new approaches is the adoption of certain critical components of conventional combination chemotherapy schedules for pediatric patients, which, when used in young and even middle-aged adults, have improved treatment outcomes. The authors of this review convincingly make the case that, when feasible, these pediatric-inspired regimens should be utilized instead of more conventional treatment plans such as hyper-CVAD (alternating cycles of [A] cyclophosphamide, doxorubicin, vincristine, and dexamethasone; and [B] high-dose methotrexate and high-dose cytarabine). There is now recognition that detection of minimal residual disease (MRD) is an important prognostic tool that should help guide therapeutic decisions. Access to sophisticated MRD immunophenotypic techniques has become an essential part of the care of patients with ALL. Finally, the genomics of ALL, although perhaps not as rich as in acute myeloid leukemia, have led to routine incorporation-and even substitution-of tyrosine kinase inhibitors in the treatment of Philadelphia chromosome (Ph)-positive ALL, the recognition of Ph-like signatures, and even investigations into other targeted agents, such as Notch inhibitors.What New Avenue of Research Seems to Hold Special Promise for ALL in the Future?Immunotherapy has emerged as the recent development with the potential for the greatest impact in ALL. The addition of the anti-CD20 antibody rituximab to induction chemotherapy has significantly improved outcomes. Inotuzumab, the antibody-drug conjugate targeting CD22, has been shown to be superior to conventional chemotherapy in inducing remissions for patients with recurrent disease. Blinatumomab, the bispecific antibody activating T cells and targeting CD19, has been used to treat both patients with active disease and those with MRD, thus permitting more patients with recurrent/refractory disease to reach hematopoietic stem cell transplantation (HSCT). Adoptive cellular therapy with T cells engineered to express chimeric antigen receptors has induced remissions even in patients who relapsed after allogeneic HSCT. Indeed, these new immunologic approaches, as well as implications of MRD detection, will lead to a re-evaluation of the role of allogeneic HSCT in ALL.Financial Disclosure:Dr. Soiffer serves as a consultant to Jazz Pharmaceuticals and Sandoz, is chair of the grant committee at Gilead, is a member of the data safety monitoring board at Juno, and is a member of the Board at Kiadis Pharma.
Intensive induction therapy has been associated with increased treatment-related mortality in older adults.[62] Thus, various attempts have been made to adjust doses of agents in order to minimize toxicity and early mortality. For example, elimination of cyclophosphamide and asparaginase resulted in reduced treatment-related mortality and improved survival in the PETHEMA ALL-96 trial.[62] Nonetheless, the German Multicenter Study Group for Adult ALL (GMALL) has successfully included reduced doses of PEG-asparaginase (500 IU/m2) in older adults with ALL, and this approach was deemed tolerable.[63] Currently, the use of Erwinia asparaginase in upfront therapy for older ALL patients (> 60 years) is being examined in a clinical trial (ClinicalTrials.gov identifier: NCT02647190), in light of the encouraging safety profile that has been reported for this agent.[64] Because of its short half-life, Erwinia asparaginase has the advantage of quick reversal upon development of toxicity. Asparaginase encapsulated within erythrocytes was developed to reduce allergic reactions and silent hypersensitivity, and its safety and ability to achieve durable asparagine depletion in older adults (≥ 55 years) was demonstrated in the phase II GRASPALL/GRAALL-SA2-2008 study.[65]
As discussed earlier, modified pediatric-inspired regimens can be used in older adults at least up to 60 years of age, but the efficacy of such regimens in the older adult population remains to be determined. Considerations for older adult regimens include eliminating asparaginase from induction and reducing its dose in consolidation, monitoring closely for vincristine and anthracycline toxicity, and careful use of high-dose methotrexate. Supportive care is critical in older adults receiving ALL therapy. This includes routine use of granulocyte colony–stimulating agents to avoid neutropenia, and routine administration of prophylactic acyclovir and antifungal therapy, including anti-Pneumocystis therapy.
It is critical to refer older adults early for allogeneic HSCT consultation, especially when adequate combination chemotherapy cannot be administered because of toxicity. Reduced-intensity conditioning allogeneic HSCT is greatly underutilized in older adults. The limited data available suggest that reduced-intensity conditioning allogeneic HSCT is well tolerated and active, resulting in a better chance of cure in older adults compared with chemotherapy alone.[66-68] Improving supportive care during transplant and introducing novel therapies prior to HSCT may further improve outcomes in the elderly. Ironically, it is possible that HSCT may have a better side-effect profile than prolonged chemotherapy in older adults.
Since reduced tolerability and chemotherapy resistance are the main factors hindering curability of ALL in older adults, novel immune-based therapies have the potential to replace or augment conventional chemotherapy in this setting, since the toxicity and activity of these novel therapies are less dependent on age and on the absence of underlying high-risk leukemia genetics. Blinatumomab has shown remarkable activity in advanced ALL, irrespective of patient age.[69] Blinatumomab is actively being tested in combination with low-dose chemotherapy and TKIs as upfront therapy in older adults with ALL.
Central Nervous System (CNS) Prophylaxis and Treatment
CNS disease at the time of ALL diagnosis is uncommon, but the risk of relapse in this sanctuary site is substantial in the absence of adequate CNS-directed prophylactic therapy. Early and frequent intrathecal administration of methotrexate in combination with intravenous chemotherapies that have sufficient blood-brain barrier penetration has emerged as the mainstay of CNS prophylaxis in modern ALL regimens, and this strategy has resulted in low risk of CNS relapse. Historically, prophylactic cranial-spinal radiation was an integral part of CNS prophylaxis regimens. However, due to prohibitive late toxicities, especially in older adults, prophylactic radiation has been largely abandoned. The prognosis of CNS involvement at diagnosis remains a matter of debate, but one study showed a worse outcome.[70] On the other hand, isolated CNS relapse portends imminent systemic relapse and mandates systemic chemotherapy in addition to CNS-directed therapy. Allogeneic HSCT consolidation after local clearance of ALL has resulted in encouraging outcomes.[71] The graft-vs-leukemia effect can potentially extend to the cerebrospinal fluid, and in one study, no clear benefit was observed for radiation-based conditioning, administration of cranial radiation prior to HSCT, or administration of post-HSCT prophylactic intrathecal chemotherapy.[71]
The Role of Allogeneic HSCT in Adults With ALL
While there is no argument against the general principle that adults with refractory and relapsed ALL should undergo allogeneic HSCT whenever feasible, how best to select candidates for consolidation with allogeneic HSCT in first remission remains a matter of debate. Precisely defining the subset of patients who will derive the maximum benefit from allogeneic HSCT in CR1 is a major challenge. While the standard recommendation is to consider allogeneic HSCT for high-risk ALL in CR1, recent data suggest a more pronounced benefit with early allogeneic HSCT in patients with standard-risk disease compared with the high-risk subset as defined by age, cytogenetics, and white blood cell count.[72,73] In addition, donor availability remains a major factor in decision making regarding the use of allogeneic HSCT. Although the advances in haploidentical HSCT have made it possible for almost every patient to have an available donor, the outcomes of transplants involving haploidentical donors are inferior when performed in relapsed/refractory disease. Therefore, it is recommended that every adult patient with ALL be offered an HSCT consultation soon after diagnosis; this is particularly important for patients with high-risk disease and those who are not treated with pediatric-type regimens.
With the use of pediatric-type regimens, minimal residual disease (MRD) has emerged as the most important factor in determining which patients should be consolidated with allogeneic HSCT.[74] Standard-risk stratification factors (age, cytogenetics, white blood cell count, etc) are less important.[75]
MRD
MRD refers to the presence of residual leukemia that is not detectable by morphology or routine flow cytometry. Both polymerase chain reaction–based and flow cytometry–based methods of MRD detection have now been standardized and are clinically available. These techniques have the ability to detect as few as 10-4 to 10-5 (1 in 10,000 to 1 in 100,000) leukemic cells. The details of MRD assessment techniques, as well as the pros and cons of each approach, are outside the scope of this article, and the reader is referred to a recent review.[76] Whichever assessment method is used, the MRD status of ALL patients in remission has emerged as a very powerful prognostic tool that trumps all other traditional risk factors. MRD is a well-established prognostic tool in children with ALL, and results are routinely used to make changes to treatment plans in this setting.
In adults, the prognostic significance of MRD data is consistent with that of pediatric data, but the exact timing of the assessment so as to be most helpful with determining whether to modify the treatment plan remains a matter of debate. The US Intergroup study showed that day-28 MRD status was prognostic, and a positive test result was associated with inferior survival.[41] MRD assessment between 4 and 16 weeks from induction has been used in various studies, and it appears that the inferior outcomes associated with MRD positivity are more pronounced in patients who do not undergo allogeneic HSCT.[77,78] Thus, it has become customary to recommend allogeneic HSCT for MRD-positive patients whenever possible, although the outcome of allogeneic HSCT in this setting is inferior to that seen in MRD-negative patients.[74] Despite the ability of intensive chemotherapy to convert MRD-positive remission to MRD-negative status before transplant in relapsed ALL, the adverse impact of initial pretransplant MRD positivity persisted after HSCT.[79] Whether the same applies to more potent immunotherapies that can achieve MRD-negative status prior to HSCT remains to be determined.
Treatment of Specific ALL Subtypes in Adults
Ph+ ALL
Ph+ ALL represents a quarter of adult ALL cases.[12] In the pre-TKI era, Ph+ ALL was associated with dismal long-term outcomes due to lower response rates and shorter durability of response with conventional chemotherapy.[80-82] The introduction of TKIs has dramatically changed the natural history of this disease, and nowadays, remission is achieved readily in the vast majority of patients.[83-86] When combined with imatinib in the GRAALL randomized study, intensified induction chemotherapy in Ph+ ALL not only did not improve OS, leukemia-free survival, or molecular response compared with reduced doses of chemotherapy, it resulted in a lower CR rate due to increased treatment-related mortality.[87] Furthermore, induction treatment with just a TKI and a corticosteroid resulted in a CR in the majority of Ph+ ALL patients, with very low toxicities despite the high median age of enrolled patients.[88,89] TKIs should be administered continuously during induction, consolidation, and maintenance therapy.[90]
The choice of TKI for newly diagnosed patients remains an open question due to the lack of randomized comparative clinical trials; however, second-generation TKIs have superior potency and wider activity in the presence of BCR-ABL1 resistance mutations. TKIs do not increase asparaginase toxicity when combined with the latter agent.[91] However, asparaginase could cause prolonged duration of high-grade hyperbilirubinemia, which may interrupt TKI therapy, especially early in the course of therapy. Thus, it is preferable to avoid asparaginase in Ph+ ALL, especially when allogeneic HSCT is planned as consolidation.
Consolidation with allogeneic HSCT remains the recommended approach for adults with Ph+ ALL who achieve remission,[87,92] even though this is no longer advocated for pediatric patients.[93] Survival rates after reduced- and full-intensity allogeneic HSCT are comparable,[94] and there is intriguing interest in consolidation with autologous HSCT in Ph+ ALL patients who achieve molecular remission.[87,95] Since relapse remains the leading cause of transplant failure, it is common practice to administer TKI maintenance for a certain period post transplant, but results with this approach have been mixed.[96,97]
T-cell ALL
The outcomes of adults with T-cell ALL have improved with modern ALL regimens, and today outcomes are comparable to those of patients with the precursor B-cell subtype.[28,41] However, the T-cell ALL subtype is consistently a negative prognostic factor for patients who relapse, and only limited salvage options are available.[98] Thus, every effort should be made to optimize upfront therapy of T-cell ALL in order to avoid the detrimental consequence of relapse.
A pediatric-inspired regimen is recommended for upfront therapy. Nelarabine has activity in relapsed T-cell ALL and is being introduced in upfront regimens. A higher dose of methotrexate (5 g/m2) has superior activity to that of lower doses in T-cell ALL/lymphoblastic lymphoma, and it is recommended in the absence of contraindications.[48] Asparaginase is another key drug that should not be eliminated in this setting. The improved outcomes in T-cell ALL seen with chemotherapy regimens have been attributed to intensive use of asparaginase, and this practice can potentially avoid the need for allogeneic HSCT in standard-risk cases.[99] Recent data have shown a high relapse rate in adults with T-cell ALL treated upfront with asparaginase-free regimens such as hyper-CVAD.[53]
Early thymic precursor (ETP) T-cell ALL is an immature subtype characterized by a unique gene expression profile and phenotype, with expression of myeloid markers. Although the ETP subtype has been associated with an inferior prognosis in adults,[100] a recent pediatric analysis showed outcomes similar to those of other T-cell ALL subtypes despite a higher rate of induction failure.[101] The expression of CD1a on leukemic blasts and lack of CD13 expression were associated with better prognosis.[28]
Treatment Approach for Relapsed/Refractory ALL in Adults, Including Novel Therapies
The prognosis of relapsed/refractory ALL is extremely poor, with 5-year OS rates as low as 7% in one study.[102] Although a morphologic remission may be achieved, this has not traditionally translated into improved survival. This may be changing with the advent of potent immunotherapeutic approaches that are capable of achieving deeper remissions (often MRD-negative) in this setting. Combination chemotherapy can result in brief remissions in a subset of patients, and for patients who attain a second remission or beyond, allogeneic HSCT should be performed whenever feasible.
Mitoxantrone-based reinduction was associated with a superior response compared with idarubicin in the randomized ALL R3 trial in children,[103] and this agent can be tried in younger adults. Clofarabine in combination with cyclophosphamide and etoposide is another tested salvage regimen, and it can achieve remission in half of adult patients with relapsed/refractory ALL.[104] This regimen can be used as a bridge to allogeneic HSCT, but the duration of remission is generally brief and toxicities may be additive with those of allogeneic HSCT. Liposomal vincristine has single-agent activity in relapsed/refractory ALL and is currently approved for adult patients with Ph− ALL in whom at least two lines of therapy have failed.[105] Nelarabine as a single agent induced CR in 36% of adult patients with relapsed/refractory T-cell ALL.[106] For patients with Ph+ ALL, second-generation TKIs, such as dasatinib and nilotinib, are active if a patient has no resistant BCR-ABL1 mutations such as T315I. Ponatinib is a third-generation TKI that can induce remission in a third of patients with relapsed/refractory Ph+ ALL, irrespective of T315I mutation status.[107]
Monoclonal antibodies
Blinatumomab is a bispecific (CD3/CD19) T-cell–engaging antibody that approximates CD3+ T cells to CD19+ B cells in order to promote the formation of immunologic synapses. This results in the release of cytokines that induce killing of CD19+ cells, independent of T-cell receptor specificity, costimulation, or peptide-antigen presentation. CD19 is universally expressed in precursor B-cell ALL. Blinatumomab has shown potent activity in morphologic relapsed/refractory ALL, as well as in MRD-positive disease, independent of chemosensitivity or previous allogeneic HSCT, and the majority of responses were MRD-negative. The CR rate was 43% in relapsed/refractory ALL and molecular response rate was as high as 80% among MRD-positive adult ALL patients.[108,109] Blinatumomab is currently approved for relapsed/refractory CD19+ ALL and can facilitate allogeneic HSCT for chemotherapy-refractory patients. In patients with MRD relapse only, blinatumomab has resulted in long-term remissions without further therapy. The main toxicities for blinatumomab include neurotoxicity, cytokine release syndrome, and B-cell depletion.
Inotuzumab is a CD22 monoclonal antibody linked to calicheamicin. CD22 is expressed in over 90% of B-cell ALL cases.[54] Inotuzumab demonstrated single-agent activity in relapsed/refractory CD22+ ALL, with an overall response rate of 58%. The antibody was tolerated but there was an increased risk of veno-occlusive disease (17%) in patients who subsequently underwent allogeneic HSCT.[110] A weekly dose was chosen for subsequent studies that was less toxic but maintained good activity. In a phase III randomized study, single-agent inotuzumab was associated with a higher CR rate (81% vs 28%; P < .001) and longer median progression-free survival (5 vs 1.8 months; P < .001) and OS (7.7 vs 6.7 months; P = .04), compared with standard intensive chemotherapy in relapsed/refractory adult ALL.[111]
Chimeric antigen receptor (CAR) T cells
CAR T cells are generated by transducing the receptor of interest in order to direct T cells toward target cells. These receptor-gene constructs typically consist of a tumor antigen–specific variable-region antibody fragment fused to the signal transduction component of the T-cell receptor and a T-cell costimulatory domain. The most successful experience so far is with CD19-directed CAR T cells, which have produced very high responses in advanced ALL, irrespective of specific CAR T-cell design.[112-114] CAR T-cell therapy requires autologous T-cell collection, followed by transduction and expansion of the T cells, and the subsequent infusion of the engineered T cells following a lymphodepleting regimen. This process can take several weeks, but the preparation time is being shortened continuously with new approaches. CAR T-cell therapy carries a risk of cytokine release syndrome, neurotoxicity, and hypogammaglobulinemia. CAR T cells have the capacity to reach various tissues that frequently are involved in ALL, including the CNS, but their persistence appears to be dependent on the specific T-cell phenotype and the costimulatory domain used in the construct.
Notch pathway inhibitors
Notch is a critical signaling pathway in tissue development, and activating mutations in various components of this pathway are present in about 60% of T-cell ALL cases. Inhibition of γ-secretase results in decreased levels of intracellular Notch 1 and downregulation of its target genes. In a phase I study of 7 patients with relapsed/refractory T-cell ALL, administration of a γ-secretase inhibitor (MK-0752) induced partial responses, but there was significant gastrointestinal toxicity.[115] Results in a xenograft model suggest that combining dexamethasone with this agent is synergistic and can potentially reduce gastrointestinal toxicity.[116] A phase I/II study of dexamethasone combined with LY3039478, a potent and selective Notch inhibitor, is actively enrolling patients (ClinicalTrials.gov identifier: NCT02518113).
Conclusion
We are at a turning point in the history of therapy for adult ALL. After decades of lagging behind the remarkable progress achieved in pediatric ALL, we finally appear to be chipping away at the once dismal outlook for adults with ALL. The use of pediatric-type regimens in more adults, the monitoring and prognosticating of disease via MRD assessment, and the use of potent immunotherapies can all be expected to contribute to improved outcomes, particularly in older patients and those with relapsed/refractory disease. The importance of referring adult ALL patients at diagnosis to specialized centers where they will have access to a broad array of novel therapies, as well as clinical trials, cannot be overemphasized.
Financial Disclosure:Dr. Pullarkat serves on advisory boards for Amgen, Baxalta, and Pfizer. Drs. Aldoss and Marcucci have no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
1. Dores GM, Devesa SS, Curtis RE, et al. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119:34-43.
2. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541-52.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30.
4. Chiaretti S, Vitale A, Cazzaniga G, et al. Clinico-biological features of 5202 patients with acute lymphoblastic leukemia enrolled in the Italian AIEOP and GIMEMA protocols and stratified in age cohorts. Haematologica. 2013;98:1702-10.
5. Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:1663-9.
6. Pullarkat ST, Danley K, Bernstein L, et al. High lifetime incidence of adult acute lymphoblastic leukemia among Hispanics in California. Cancer Epidemiol Biomarkers Prev. 2009;18:611-5.
7. Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45:1494-8.
8. Perez-Andreu V, Roberts KG, Xu H, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125:680-6.
9. Pullarkat V, Slovak ML, Kopecky KJ, et al. Impact of cytogenetics on the outcome of adult acute lymphoblastic leukemia: results of Southwest Oncology Group 9400 study. Blood. 2008;111:2563-72.
10. Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11:429-38.
11. Moorman AV, Harrison CJ, Buck GA, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189-97.
12. Moorman AV, Chilton L, Wilkinson J, et al. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood. 2010;115:206-14.
13. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005-15.
14. Herold T, Baldus CD, Gokbuget N. Ph-like acute lymphoblastic leukemia in older adults. N Engl J Med. 2014;371:2235.
15. Kobayashi K, Miyagawa N, Mitsui K, et al. TKI dasatinib monotherapy for a patient with Ph-like ALL bearing ATF7IP/PDGFRB translocation. Pediatr Blood Cancer. 2015;62:1058-60.
16. Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243-6.
17. Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312-21.
18. Yoda A, Yoda Y, Chiaretti S, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107:252-7.
19. Chiaretti S, Brugnoletti F, Messina M, et al. CRLF2 overexpression identifies an unfavourable subgroup of adult B-cell precursor acute lymphoblastic leukemia lacking recurrent genetic abnormalities. Leuk Res. 2016;41:36-42.
20. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470-80.
21. Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123:3739-49.
22. Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. 2009;27:5202-7.
23. Aldoss I, Dagis A, Palmer J, et al. Therapy-related ALL: cytogenetic features and hematopoietic cell transplantation outcome. Bone Marrow Transplant. 2015;50:746-8.
24. Ganzel C, Devlin S, Douer D, et al. Secondary acute lymphoblastic leukaemia is constitutional and probably not related to prior therapy. Br J Haematol. 2015;170:50-5.
25. Tang G, Zuo Z, Thomas DA, et al. Precursor B-acute lymphoblastic leukemia occurring in patients with a history of prior malignancies: Is it therapy-related? Haematologica. 2012;97:919-25.
26. Hayette S, Tigaud I, Maguer-Satta V, et al. Recurrent involvement of the MLL gene in adult T-lineage acute lymphoblastic leukemia. Blood. 2002;99:4647-9.
27. Rubnitz JE, Camitta BM, Mahmoud H, et al. Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol. 1999;17:191-6.
28. Marks DI, Paietta EM, Moorman AV, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114:5136-45.
29. Neumann M, Coskun E, Fransecky L, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS One. 2013;8:e53190.
30. Harrison CJ, Moorman AV, Broadfield ZJ, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:552-9.
31. Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26:123-35.
32. Larson RA, Dodge RK, Linker CA, et al. A randomized controlled trial of filgrastim during remission induction and consolidation chemotherapy for adults with acute lymphoblastic leukemia: CALGB study 9111. Blood. 1998;92:1556-64.
33. Giebel S, Thomas X, Hallbook H, et al. The prophylactic use of granulocyte-colony stimulating factor during remission induction is associated with increased leukaemia-free survival of adults with acute lymphoblastic leukaemia: a joint analysis of five randomised trials on behalf of the EWALL. Eur J Cancer. 2012;48:360-7.
34. Bhatia S, Landier W, Hageman L, et al. Systemic exposure to thiopurines and risk of relapse in children with acute lymphoblastic leukemia: a Children’s Oncology Group study. JAMA Oncol. 2015;1:287-95.
35. Boissel N, Auclerc MF, Lheritier V, et al. Should adolescents with acute lymphoblastic leukemia be treated as old children or young adults? Comparison of the French FRALLE-93 and LALA-94 trials. J Clin Oncol. 2003;21:774-80.
36. de Bont JM, Holt B, Dekker AW, et al. Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia. 2004;18:2032-5.
37. Ramanujachar R, Richards S, Hann I, et al. Adolescents with acute lymphoblastic leukaemia: outcome on UK national paediatric (ALL97) and adult (UKALL XII/E2993) trials. Pediatr Blood Cancer. 2007;48:254-61.
38. Stock W, La M, Sanford B, et al. What determines the outcomes for adolescents and young adults with acute lymphoblastic leukemia treated on cooperative group protocols? A comparison of Children’s Cancer Group and Cancer and Leukemia Group B studies. Blood. 2008;112:1646-54.
39. Douer D, Aldoss I, Lunning MA, et al. Pharmacokinetics-based integration of multiple doses of intravenous pegaspargase in a pediatric regimen for adults with newly diagnosed acute lymphoblastic leukemia. J Clin Oncol. 2014;32:905-11.
40. DeAngelo DJ, Stevenson KE, Dahlberg SE, et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia. 2015;29:526-34.
41. Stock W, Luger SM, Advani AS, et al. Favorable outcomes for older adolescents and young adults (AYA) with acute lymphoblastic leukemia (ALL): early results of U.S. intergroup trial C10403. Blood. 2014;124:abstr 796.
42. Seftel MD, Neuberg D, Zhang MJ, et al. Pediatric-inspired therapy compared to allografting for Philadelphia chromosome-negative adult ALL in first complete remission. Am J Hematol. 2016;91:322-9.
43. Ribera JM, Oriol A, Sanz MA, et al. Comparison of the results of the treatment of adolescents and young adults with standard-risk acute lymphoblastic leukemia with the Programa Espanol de Tratamiento en Hematologia pediatric-based protocol ALL-96. J Clin Oncol. 2008;26:1843-9.
44. Rijneveld AW, van der Holt B, Daenen SM, et al. Intensified chemotherapy inspired by a pediatric regimen combined with allogeneic transplantation in adult patients with acute lymphoblastic leukemia up to the age of 40. Leukemia. 2011;25:1697-703.
45. Huguet F, Leguay T, Raffoux E, et al. Pediatric-inspired therapy in adults with Philadelphia chromosome-negative acute lymphoblastic leukemia: the GRAALL-2003 study. J Clin Oncol. 2009;27:911-8.
46. Storring JM, Minden MD, Kao S, et al. Treatment of adults with BCR-ABL negative acute lymphoblastic leukaemia with a modified paediatric regimen. Br J Haematol. 2009;146:76-85.
47. Larsen EC, Devidas M, Chen S, et al. Dexamethasone and high-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: a report from Children’s Oncology Group study AALL0232. J Clin Oncol. 2016 Apr 25. [Epub ahead of print]
48. Asselin BL, Devidas M, Wang C, et al. Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children’s Oncology Group (POG 9404). Blood. 2011;118:874-83.
49. Sakura T, Hayakawa F, Sugiura I, et al. Effectiveness of high-dose MTX therapy for adult Ph-negative ALL by randomized trial: JALSG ALL202-O. Blood. 2015;126:abstr 79.
50. Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101:2788-801.
51. Morris K, Weston H, Mollee P, et al. Outcome of treatment of adult acute lymphoblastic leukemia with hyperfractionated cyclophosphamide, doxorubicin, vincristine, dexamethasone/methotrexate, cytarabine: results from an Australian population. Leuk Lymphoma. 2011;52:85-91.
52. Buyukasik Y, Acar K, Kelkitli E, et al. Hyper-CVAD regimen in routine management of adult acute lymphoblastic leukemia: a retrospective multicenter study. Acta Haematol. 2013;130:199-205.
53. Kozlowski P, Astrom M, Ahlberg L, et al. High relapse rate of T cell acute lymphoblastic leukemia in adults treated with Hyper-CVAD chemotherapy in Sweden. Eur J Haematol. 2014;92:377-81.
54. Raponi S, De Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52:1098-107.
55. Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28:3880-9.
56. Maury S, Chevret S, Thomas X, et al. Addition of rituximab improves the outcome of adult patients with CD20-positive, Ph-negative, B-cell precursor acute lymphoblastic leukemia (BCP-ALL): results of the randomized GRAALL-R 2005 study. Blood. 2015;126:abstr 1.
57. Stock W, Douer D, DeAngelo DJ, et al. Prevention and management of asparaginase/PEGasparaginase-associated toxicities in adults and older adolescents: recommendations of an expert panel. Leuk Lymphoma. 2011;52:2237-53.
58. Aldoss I, Douer D, Behrendt CE, et al. Toxicity profile of repeated doses of PEG-asparaginase incorporated into a pediatric-type regimen for adult acute lymphoblastic leukemia. Eur J Haematol. 2016;96:375-80.
59. Mitchell L, Andrew M, Hanna K, et al. Trend to efficacy and safety using antithrombin concentrate in prevention of thrombosis in children receiving L-asparaginase for acute lymphoblastic leukemia. Results of the PAARKA study. Thromb Haemost. 2003;90:235-44.
60. Hunault-Berger M, Chevallier P, Delain M, et al. Changes in antithrombin and fibrinogen levels during induction chemotherapy with L-asparaginase in adult patients with acute lymphoblastic leukemia or lymphoblastic lymphoma. Use of supportive coagulation therapy and clinical outcome: the CAPELAL study. Haematologica. 2008;93:1488-94.
61. Sive JI, Buck G, Fielding A, et al. Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG 2993 trial. Br J Haematol. 2012;157:463-71.
62. Sancho JM, Ribera JM, Xicoy B, et al. Results of the PETHEMA ALL-96 trial in elderly patients with Philadelphia chromosome-negative acute lymphoblastic leukemia. Eur J Haematol. 2007;78:102-10.
63. Gökbuget N. How I treat older patients with ALL. Blood. 2013;122:1366-75.
64. Plourde PV, Jeha S, Hijiya N, et al. Safety profile of asparaginase Erwinia chrysanthemi in a large compassionate-use trial. Pediatr Blood Cancer. 2014;61:1232-8.
65. Hunault-Berger M, Leguay T, Huguet F, et al. A phase 2 study of L-asparaginase encapsulated in erythrocytes in elderly patients with Philadelphia chromosome negative acute lymphoblastic leukemia: The GRASPALL/GRAALL-SA2-2008 study. Am J Hematol. 2015;90:811-8.
66. Stein AS, Palmer JM, O’Donnell MR, et al. Reduced-intensity conditioning followed by peripheral blood stem cell transplantation for adult patients with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2009;15:1407-14.
67. Kanamori H, Mizuta S, Kako S, et al. Reduced-intensity allogeneic stem cell transplantation for patients aged 50 years or older with B-cell ALL in remission: a retrospective study by the Adult ALL Working Group of the Japan Society for Hematopoietic Cell Transplantation. Bone Marrow Transplant. 2013;48:1513-8.
68. Mohty M, Labopin M, Volin L, et al. Reduced-intensity versus conventional myeloablative conditioning allogeneic stem cell transplantation for patients with acute lymphoblastic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation. Blood. 2010;116:4439-43.
69. Kantarjian HM, Stein AS, Bargou RC, et al. Blinatumomab treatment of older adults with relapsed/refractory B-precursor acute lymphoblastic leukemia: results from two phase 2 studies. Cancer. 2016 May 3. [Epub ahead of print]
70. Lazarus HM, Richards SM, Chopra R, et al. Central nervous system involvement in adult acute lymphoblastic leukemia at diagnosis: results from the international ALL trial MRC UKALL XII/ECOG E2993. Blood. 2006;108:465-72.
71. Aldoss I, Al Malki MM, Stiller T, et al. Implications and management of central nervous system involvement before allogeneic hematopoietic cell transplantation in acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2016;22:575-8.
72. Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111:1827-33.
73. Cornelissen JJ, van der Holt B, Verhoef GE, et al. Myeloablative allogeneic versus autologous stem cell transplantation in adult patients with acute lymphoblastic leukemia in first remission: a prospective sibling donor versus no-donor comparison. Blood. 2009;113:1375-82.
74. Bar M, Wood BL, Radich JP, et al. Impact of minimal residual disease, detected by flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia. Leuk Res Treatment. 2014;2014:421723.
75. Aldoss I, Tsai NC, Slovak ML, et al. Cytogenetics does not impact outcomes in adult patients with acute lymphoblastic leukemia undergoing allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016 Apr 1. [Epub ahead of print]
76. van Dongen JJ, van der Velden VH, Bruggemann M, et al. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125:3996-4009.
77. Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120:1868-76.
78. Dhedin N, Huynh A, Maury S, et al. Role of allogeneic stem cell transplantation in adult patients with Ph-negative acute lymphoblastic leukemia. Blood. 2015;125:2486-96.
79. Eckert C, Chen-Santel C, Peters C, et al. Interventional intensification of chemotherapy prior to hematopoietic stem cell transplantation reduces residual leukemia but does not improve survival in children with relapsed acute lymphoblastic leukemia. Blood. 2014;124:abstr 61.
80. Dombret H, Gabert J, Boiron JM, et al. Outcome of treatment in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia-results of the prospective multicenter LALA-94 trial. Blood. 2002;100:2357-66.
81. Gleissner B, Gökbuget N, Bartram CR, et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood. 2002;99:1536-43.
82. Fielding AK, Rowe JM, Buck G, et al. UKALL XII/ECOG 2993: addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123:843-50.
83. Lee KH, Lee JH, Choi SJ, et al. Clinical effect of imatinib added to intensive combination chemotherapy for newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia. 2005;19:1509-16.
84. de Labarthe A, Rousselot P, Huguet-Rigal F, et al. Imatinib combined with induction or consolidation chemotherapy in patients with de novo Philadelphia chromosome-positive acute lymphoblastic leukemia: results of the GRAAPH-2003 study. Blood. 2007;109:1408-13.
85. Towatari M, Yanada M, Usui N, et al. Combination of intensive chemotherapy and imatinib can rapidly induce high-quality complete remission for a majority of patients with newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia. Blood. 2004;104:3507-12.
86. Yanada M, Takeuchi J, Sugiura I, et al. High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol. 2006;24:460-6.
87. Chalandon Y, Thomas X, Hayette S, et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood. 2015;125:3711-9.
88. Vignetti M, Fazi P, Cimino G, et al. Imatinib plus steroids induces complete remissions and prolonged survival in elderly Philadelphia chromosome-positive patients with acute lymphoblastic leukemia without additional chemotherapy: results of the Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA) LAL0201-B protocol. Blood. 2007;109:3676-8.
89. Foa R, Vitale A, Vignetti M, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118:6521-8.
90. Wassmann B, Pfeifer H, Goekbuget N, et al. Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2006;108:1469-77.
91. Goekbuget N, Baumann A, Beck J, et al. PEG-asparaginase intensification in adult acute lymphoblastic leukemia (ALL): significant improvement of outcome with moderate increase of liver toxicity in the German Multicenter Study Group for Adult ALL (GMALL) study 07/2003. Blood. 2010;116:abstr 494.
92. Fielding AK, Rowe JM, Richards SM, et al. Prospective outcome data on 267 unselected adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia confirms superiority of allogeneic transplantation over chemotherapy in the pre-imatinib era: results from the International ALL Trial MRC UKALL XII/ECOG 2993. Blood. 2009;113:4489-96.
93. Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28:1467-71.
94. Bachanova V, Marks DI, Zhang MJ, et al. Ph+ ALL patients in first complete remission have similar survival after reduced intensity and myeloablative allogeneic transplantation: impact of tyrosine kinase inhibitor and minimal residual disease. Leukemia. 2014;28:658-65.
95. Wetzler M, Watson D, Stock W, et al. Autologous transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia achieves outcomes similar to allogeneic transplantation: results of CALGB Study 10001 (Alliance). Haematologica. 2014;99:111-5.
96. Brissot E, Labopin M, Beckers MM, et al. Tyrosine kinase inhibitors improve long-term outcome of allogeneic hematopoietic stem cell transplantation for adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia. Haematologica. 2015;100:392-9.
97. Pfeifer H, Wassmann B, Bethge W, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia. 2013;27:1254-62.
98. Nguyen K, Devidas M, Cheng SC, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22:2142-50.
99. Hoelzer D, Thiel E, Arnold R, et al. Successful subtype oriented treatment strategies in adult T-ALL; results of 744 patients treated in three consecutive GMALL studies. Blood. 2009;114:abstr 324.
100. Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127:1863-9.
101. Wood BL, Winter SS, Dunsmore KP, et al. T-lymphoblastic leukemia (T-ALL) shows excellent outcome, lack of significance of the early thymic precursor (ETP) immunophenotype, and validation of the prognostic value of end-induction minimal residual disease (MRD) in Children’s Oncology Group (COG) study AALL0434. Blood. 2014;124:abstr 1.
102. Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944-50.
103. Parker C, Waters R, Leighton C, et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet. 2010;376:2009-17.
104. Huguet F, Leguay T, Raffoux E, et al. Clofarabine for the treatment of adult acute lymphoid leukemia: the Group for Research on Adult Acute Lymphoblastic Leukemia intergroup. Leuk Lymphoma. 2015;56:847-57.
105. O’Brien S, Schiller G, Lister J, et al. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult Philadelphia chromosome-negative acute lymphoblastic leukemia. J Clin Oncol. 2013;31:676-83.
106. Gökbuget N, Basara N, Baurmann H, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. 2011;118:3504-11.
107. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783-96.
108. Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57-66.
109. Topp MS, Kufer P, Gökbuget N, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. 2011;29:2493-8.
110. Kantarjian H, Thomas D, Jorgensen J, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119:2728-36.
111. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016 Jun 12. [Epub ahead of print]
112. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25.
113. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517-28.
114. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507-17.
115. Deangelo DJ, Stone RM, Silverman LB, et al. A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol. 2006;24(suppl 18):abstr 6585.
116. Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23:1374-7.
117. Moorman AV. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016;101:407-16.
118. Testi AM, Valsecchi MG, Conter V, et al. Difference in outcome of adolescents with acute lymphoblastic leukemia (ALL) enrolled in pediatric (AIEOP) and adult (GIMEMA) protocols. Blood. 2004;104:1954.
119. Boissel N, Sender LS. Best practices in adolescent and young adult patients with acute lymphoblastic leukemia: a focus on asparaginase. J Adolesc Young Adult Oncol. 2015;4:118-28.