Cancer Metabolism as a Therapeutic Target
Ongoing studies are attempting to understand the reasons that tumor cells engage in aerobic glycolysis in lieu of oxidative phosphorylation. In this review, we discuss known benefits to tumor cells from this metabolic switch, and we highlight key enzymes that play a role in aerobic glycolysis. We also describe novel therapeutic options targeting glucose metabolism.
Cancer is now recognized to be a disease arising from both genetic and metabolic abnormalities. In the mid-1900s, Otto Warburg described the phenomenon of elevated glucose consumption and aerobic glycolysis, and the dependence of cancer cells on this phenomenon for proliferation and growth. The Warburg effect has formed the basis of such diagnostic and prognostic imaging modalities as positron emission tomography (PET); however, we have not yet capitalized on this phenomenon for therapy. Several mechanisms have now been shown to contribute to the Warburg effect.
Ongoing studies are attempting to understand the reasons that tumor cells engage in aerobic glycolysis in lieu of oxidative phosphorylation, and the advantages that accrue to them as a result. In this review, we discuss known benefits to tumor cells from this metabolic switch, and we highlight key enzymes that play a role in aerobic glycolysis. We also describe novel therapeutic options targeting glucose metabolism and the importance of continuing to understand the metabolic plasticity of cancer.
Introduction
Treatment of cancer has undergone several evolutionary changes in the last decade as discoveries of various pathways leading to oncogenesis have continued to unfold. Despite these advances, cancer deaths have decreased by only a little more than 1% over the last 10 years.[1] A total of 1,638,910 new cancer cases and 577,190 deaths from cancer were projected to occur in the United States in 2012.[1] One of the consistently encountered hurdles in the treatment of all cancers has been development of resistance to currently available treatment modalities. Thus, there is a need to develop newer treatment modalities to combat ensuing resistance and to improve survival in affected patients.
The Warburg Effect
Over the last century, cancer therapy has focused on understanding and targeting the genetic basis of tumor development and progression. While this approach has led to significant advances in personalized cancer therapy, the problem of how to target tumor cells that revert to alternative pathways to sustain survival and proliferation remains a stumbling block. The genetic heterogeneity of cancer in itself suggests the need for broader approaches that can target the wide range of epigenetic and genetic phenomena fueling cancer growth.
Cancer is also a disease of altered metabolism. This phenomenon, recognized close to 100 years ago, is now accepted as one of the fundamental hallmarks of cancer.[2] Targeting altered cancer metabolism may, in fact, provide us with greater opportunities to target genetic heterogeneity within any particular cancer.
Normal cells generate adenosine triphosphate (ATP) primarily by oxidation of glucose in the presence of oxygen via the highly efficient mitochondrial oxidative phosphorylation (OXPHOS) pathway. In contrast, tumor cells rely on the inefficient glycolytic pathway for generation of ATP, even in the presence of oxygen, necessitating increased rates of glucose consumption to maintain energy production.[3] This phenomenon, termed aerobic glycolysis, was first described by Otto Warburg in the mid-1900s and was originally thought to be a result of defects in oxidative phosphorylation.[4] More recent studies have shown that tumor cells do contain functional mitochondria,[5] yet they still produce excessive amounts of lactate, suggesting that the enhanced glycolytic flux may confer a growth advantage. In support of this notion, tumor cells forced to revert to OXPHOS by RNAi-mediated suppression of lactate dehydrogenase A (LDH-A; the enzyme responsible for conversion of pyruvate to lactate)[5] or by chemical inhibition of LDH-A[6] demonstrate reduced proliferation. Additionally, tumor cells treated with dichloroacetate (DCA), which inhibits pyruvate dehydrogenase kinase (PDK) and increases mitochondrial metabolism of pyruvate, demonstrate reduced rates of proliferation.[7] These and other studies have now stimulated a renewed interest in therapies targeting glycolytic metabolism.
FIGURE 1


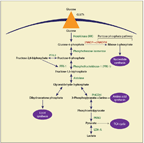
The Impact of Glucose on the Synthesis of Nucleotides, Amino Acids, Fatty Acids, and ATP-and on Redox Homeostasis
The ATP required for growth is mainly obtained through inefficient glycolysis; this occurs at an increased rate, which is necessary to sustain the high rate of cell proliferation seen in growing tumors.[10,11] It has been suggested that with an unlimited glucose supply, glycolysis is capable of producing energy at a much faster rate than OXPHOS.[3] There is emerging literature to suggest that this switch to aerobic glycolysis from mitochondrial respiration also occurs to support anabolic growth.[8] Glucose, in addition to being a source of ATP, can also serve as a precursor in a variety of biosynthetic pathways necessary for duplication of cell mass prior to cell division. Glucose-derived carbon is used to generate intermediates for nonessential amino acid synthesis, nucleotide synthesis, and fatty acid synthesis.[9] Besides, many intermediate metabolites of glycolysis-such as glucose-6-phosphate, 3-phosphoglycerate, phosphoenolpyruvate, and pyruvate-are key precursors in the biosynthesis of several amino acids required for the growth of cancer cells.[12] Figure 1 shows the impact of glucose on the synthesis of nucleotides, amino acids, fatty acids, and ATP-and on redox homeostasis in normal cells.
It was also proposed recently that persistent activation of glycolysis creates a state of metabolic acidosis that is toxic to normal cells through the p53-dependent apoptosis pathway triggered by increased caspase activity-but that is not toxic to cancer cells, presumably because of mutations in p53 or other components of the apoptotic pathway.[13,14] It has now been consistently shown that tumor cells exhibit maximal growth rates in a relatively acidic medium (~pH 6.8).[15] This acidic environment has also been shown to support acquisition of angiogenesis and the ability to invade, both of which are hallmarks of cancer cells.[16] The concept that cancer cells have an increased dependence on glycolysis has been further strengthened by the observation that most glycolysis inhibitors, such as oxamate, DCA, and PDK1 siRNA, induce apoptosis in cancers such as multiple myeloma.[17] Some of these inhibitors, eg, DCA when combined with bortezomib (Velcade), have shown additive cytotoxic effects.[17]
Several mechanisms have now been shown to contribute to the Warburg effect. Oncogenes and transcription factors, such as AKT, C-MYC, RAS, p53, and HIF1, upregulate various components of the glycolytic pathway,[18] thus facilitating the glycolytic shift. The hypoxic microenvironment associated with the preangiogenic phase of tumor development promotes stabilization of HIF1 that can induce expression of glucose transporter 1 (GLUT1), LDH-A, C-MYC[19] and PDK3,[20] facilitating aerobic glycolysis. A switch to the embryonic isoform of pyruvate kinase (PK)-ie, PKM2-is also thought to enhance anabolic processes dependent on glycolytic intermediates, thereby facilitating tumor cell proliferation,[21] while overexpression of hexokinase (HK) II in various cancers also facilitates glycolysis.[22]
The genetic and epigenetic changes in a cancer cell that contribute to the glycolytic phenotype also contribute to the progressive development of resistance to chemotherapeutics that in part may be a consequence of increased glucose catabolism. This is evident from studies demonstrating that stromal cell co-culture of leukemia cells induces the expression of uncoupling proteins that promote the switch to aerobic glycolysis and an accompanying induction of chemoresistance.[23] In addition, the switch to glycolysis can promote chemoresistance by suppressing activation of BAX and BAD, pro-apoptotic signaling effectors that are regulated by glucose.[24,25]
FIGURE 2



PET Scan Showing Numerous Lesions With Active Metabolic Uptake in a Patient With Large B-Cell Lymphoma of the Skin
In sum, altered metabolism, including increased glycolysis with enhanced glucose consumption, is consistently observed in various cancers. The Warburg effect has formed the basis of positron emission tomography (PET) scanning with fluorine-18 fluorodeoxyglucose (18F-FDG), which is now increasingly being used in the diagnosis and prognosis of various malignancies. PET exploits the increased glucose uptake by malignant cells compared with most normal cells; FDG uptake is, therefore, directly proportional to tumor burden.[26] Figure 2 shows a PET scan of a patient with large B-cell lymphoma of the skin; the scan demonstrates numerous active metabolic lesions with increased uptake of glucose. Two large independent clinical myeloma studies demonstrated the early appearance of PET-positive lesions (in 76% of 192 patients tested), indicating an early reliance on elevated glucose metabolism[27,28]; more importantly, both studies demonstrated that the persistence and extent of PET positivity correlated with lower event-free survival,[27,28] underscoring the usefulness of targeting aberrant glucose utilization in myeloma.
Previous studies have demonstrated the utility of caloric restriction in inducing chemosensitization to a wide range of chemotherapeutics. It was shown that starvation induced sensitization to radio- or chemotherapy and led to extended survival in an in vivo glioma model of astrocytoma and glioblastoma multiforme.[29] The same phenomenon was also demonstrated in a mouse neuroblastoma model in which fasting cycles and chemotherapy together resulted in long-term cancer-free survival.[30] The same group went on to explore 4T1 breast cancer cells, in which it was seen that short-term starvation resulted in increased phosphorylation of the stress-sensitizing Akt and S6 kinases, and increased oxidative stress, caspase-3 cleavage, DNA damage, and apoptosis.[30]
Given the diagnostic and prognostic usefulness of PET, there is a great potential for cancer therapeutics that target glucose utilization. The obstacle to targeting altered glucose metabolism is the identification of tumor-specific rate-limiting steps-and importantly, the metabolic plasticity that cells may engage in to bypass inhibition of these rate-limiting steps.
Targeting Glucose Metabolism
Recent research has focused on ways to target the increased dependence of cancer cells on glycolysis, with the objective of developing new chemotherapeutic and chemosensitizing treatments. Several glycolytic targets are currently being explored. This list includes targets such as HK,[31] 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3),[32] and PDK4,[33] with newer investigations exploring such avenues as enzymes like PKM2, an isoenzyme of pyruvate kinase.[34] PKM2 exhibits lower enzymatic activity compared to PKM1 and is normally expressed in embryonic and proliferating cells. However, tumor cells switch to expression of the PKM2 isoform that promotes the Warburg effect and glycolysis, in part by constraining oxidative metabolism,[35] as well as by translocating to the nucleus to promote the expression of genes associated with tumor cell proliferation.[36] Several compounds that target HK have been developed and tested, including 3-bromopyruvate, lonidamine, and 2-deoxyglucose.[37-39] However, dose-limiting toxicities and administration issues have prevented these compounds from moving ahead in clinical trials.
Targeting the Enzymes That Regulate Glycolysis
The key enzymes catalyzing three rate-limiting steps in glycolysis are HK (or glucokinase), phosphofructokinase (PFK), and PK. These enzymes are upregulated by insulin and downregulated by glucagon. Recent research has focused on targeting these enzymes and other steps in glycolysis in order to develop target-specific treatments for various malignancies.
One of the well-researched targets for the possible development of cancer therapy has been HK, which regulates the first step of glycolysis. It has been shown that malignant cells overexpress HKII, an isoform of HK. The HKII expressed by malignant cells is associated with the outer mitochondrial membrane protein VDAC (voltage-dependent anion channel).[40] Many preclinical studies have used small-molecule compounds such as 3-bromopyruvate (3-BP) to target HKII.[41]
It has also been shown that cancer cells overexpress the M2 isoform of the tightly regulated enzyme PK (PKM2), which is another enzyme that controls glycolytic flux.[34] There are data to suggest that inhibiting or activating PKM2 in cancer cells inhibits tumor growth.[34]
In addition, some recent studies have described a mechanistic relationship between PKM2 activity in cells and serine biosynthesis.[41] Serine can bind to and activate human PKM2, and PKM2 activity in cells is reduced in response to serine deprivation.
Phosphoglycerate dehydrogenase (PHGDH) converts 3-phosphoglycerate into 3-phosphohydroxypyruvate. This is the first step in serine biosynthesis and a rate-limiting step. A few subsets of human melanoma and breast cancers have been found to have high levels of PHGDH, and these cancer cells are dependent on these enzymes for growth.[42]
One of the crucial steps in glycolysis is the reduction of pyruvate to lactate. During this step, the cytosolic NADH/NAD+ (nicotinamide adenine dinucleotide hydride/nicotinamide adenine dinucleotide) ratio is high. This reaction permits the regeneration of NAD+, needed as an electron acceptor to maintain cytosolic glucose catabolism. This redox reaction is catalyzed by LDH-A. Inhibition of this enzyme as a potential way to inhibit glycolysis has been explored by many researchers. Fantin et al found that LDH-A inhibition in neu-initiated mammary epithelial tumor cell lines resulted in enhanced OXPHOS. Some later studies have demonstrated that LDH-A knockdown causes significant reduction in metastatic potential in a xenograft mouse model of hepatocellular carcinoma.[43]
The tricarboxylic acid (TCA) cycle (Krebs cycle) is a core pathway for the metabolism of sugars, lipids, and amino acids. It takes place in the mitochondria and involves the oxidation of the acetyl moiety of acetyl-coenzyme A to CO2, generating NADH and flavin adenine dinucleotide hydride (FADH2), whose electrons fuel the mitochondrial respiratory chain for ATP generation. The TCA cycle is also a major pathway for interconversion of metabolites arising from transamination and deamination of amino acids; it provides the substrates for amino acid synthesis by transamination, as well as for gluconeogenesis and fatty acid synthesis. It has been suggested that inherited and acquired alterations in TCA cycle enzymes, including succinate dehydrogenase, fumarate hydratase, and isocitrate dehydrogenase (IDH), can contribute to tumorigenesis. There are some preclinical data suggesting that IDH genes 1 and 2 (IDH1, IDH2) are mutated in cancer cells. It has been shown that 2-hydroxyglutarate production by mutated enzymes is linked to cancer pathogenesis. Decreased expression of wild-type enzyme using RNAi can impair the proliferation of wild-type IDH-expressing cancer cells.[51]
Targeting Glucose Transporters
The facilitative transport of glucose via glucose transporters into the cell is a key rate-limiting step in glucose metabolism. Glucose transporters on the cell surface and the affinity of transporters for glucose control this step.
There are two main classes of hexose transporters that are used to transport glucose into the cell; the first class is referred to as the sodium-dependent glucose transporter (SGLT) family and the second class is the glucose transporter (GLUT) family.[44,45] The fundamental difference between these two classes of hexose transporters is that the GLUTs act as facilitative transporters of glucose along a concentration gradient while the SGLTs employ the sodium-electrochemical gradient to transport glucose across the concentration gradient.
The genes that control the expression of GLUTs have the gene symbol SLC2A, since the genes belong to the 2A solute carrier family. At present, 14 isoforms of the GLUT genes have been described, and these genes have been grouped into three classes.[44,45] Class I GLUT genes were the first group of GLUT genes to be described, and are comprised of GLUT1-4 and GLUT14 (thought to be a gene duplicate of GLUT3). The most commonly studied of this group of genes are GLUT1 and GLUT4. GLUT1 is found to be overexpressed in a broad assortment of malignancies, including, but not limited to, hepatocellular cancer, gastrointestinal stromal tumor, diffuse large B-cell lymphoma, and colorectal cancer.[44,45] It has been postulated that GLUT1 overexpression correlates with tumor grade, cancer stage, and prognosis.
Class II GLUTs are made up of GLUTs 5, 7, 9, and 11, and mainly are involved in the transportation of fructose. Of these GLUTs, GLUT5 has been proposed as playing a major role in tumor proliferation, with overexpression in certain cancers compared with normal cells. This property has led to the investigation of 6–deoxy-6-[18F] fluoro-D-fructose as an alternative to [18F]-FDG-PET as a radiotracer in certain cancers in which [18F]-FDG-PET cannot be used.[30,31] Class III GLUTs are the least described group of GLUTs, and comprise GLUTs 6, 8, 10, and 12, as well as GLUT13 (also known as H+ myoinositol transporter).[44,45]
Knowledge of glucose transport in cancer cells is important in cancer therapeutics development. This knowledge is currently being used by investigators to explore the role of GLUT1 inhibitors in the treatment of cancer. It has been shown that renal cell carcinomas (RCCs) that exhibit loss of the von Hippel-Lindau (VHL) tumor suppressor gene overexpress GLUT1.[46] This study also identified a series of compounds, the 3-series, exemplified by STF-31, which has the potential to inhibit the growth of RCCs by directly binding to GLUT1 and impeding glucose transport across these tumors without affecting the normal tissue.[46] Moreover, the study showed that the activity of these compounds in the tumor could be monitored by [18F]-FDG uptake via micro-PET imaging, further strengthening the evidence for the role of glucose metabolism in the development of these tumors.
Another, widely studied series of compounds targeting the glucose receptors are the HIV-1 protease inhibitors ritonavir (Norvir) and indinavir (Crixivan). HIV-1 protease inhibitors are capable of inhibiting GLUT4 in Xenopus laevis oocytes.[47] This effect has been postulated to be responsible for various metabolic side effects caused by these drugs, including peripheral lipodystrophy, hyperlipidemia, insulin resistance, and in some cases, overt type 2 diabetes. This finding was further verified in mouse models using mice lacking the GLUT4 transporters.[48] It was found that ritonavir reduced peripheral insulin sensitivity in control mice but not in the mice lacking GLUT4 transporters. Serum insulin levels were also reduced in ritonavir-treated mice.[48] Based on these observations, recent studies have used this off-target effect of ritonavir to reduce myeloma proliferation and viability and increase chemosensitivity.[48] It was also shown that this effect of ritonavir on the growth of multiple myeloma cells was mediated through GLUT4 inhibition. Ritonavir has also demonstrated efficacy in ovarian cancer, lung cancer, and breast cancer, although the exact pathway of its mechanism of action in these malignancies still remains to be elucidated.
Targeting Compensatory Metabolism
Inhibitors that target glucose metabolism through glycolysis, which is the main source of energy for rapidly proliferating cancer cells, can cause metabolic reprogramming of cancer cells, resulting in a switch to alternative modes of metabolism to sustain growth. Some of the important alternative metabolic pathways include the TCA cycle, glutamine metabolism, and fatty acid metabolism.
Glutamine’s involvement in oxidative mitochondrial metabolism in cancer cells was reported as early as the 1970s.[49] Some recent data have shown that cancer cells can switch carbon sources between glutamine and glucose, depending on nutrient availability. In glioblastoma cells, it was shown that glucose deprivation caused a large increase in the activity of glutamate dehydrogenase (GDH), and GDH was shown to be required for cells to survive impairments in glycolysis. Reciprocally, impairment of glutamine metabolism led cells to adapt to become fully reliant on glucose for mitochondrial metabolism.[50]
There are preclinical data to suggest that glutaminase 1 (GLS1) converts glutamine to glutamate, and that GDH converts glutamate to α-keto glutarate as a source of anapleurotic carbon for the TCA cycle. GDH is required for proliferation of some cells; inhibition of GLS1 impairs proliferation of some cells. This hypothesis has been tested in several in vitro models.[51]
Recent research has focused on exploring the use of metformin to target mitochondrial complex 1. The bi-
guanide metformin, a widely used drug for the treatment of type 2 diabetes, may exert cancer chemopreventive effects by suppressing the transformative and hyperproliferative processes that initiate carcinogenesis. Epidemiologic studies have correlated metformin use with a reduced risk of cancer in patients with diabetes, earning the drug recognition as a possible antineoplastic agent for various types of malignancies.[52] Bowker et al also reported that diabetic patients who had been treated with metformin for a period of at least 5 to 5.5 years had a lower overall mortality and a lower mortality from cancer (3.5% vs 4.9% in diabetic patients who did not receive metformin at all).
The effect of metformin in cancer treatment is postulated to be associated with both direct effects of the drug and indirect effects. Metformin is known to activate 5? adenosine monophosphate-activated kinase (AMPK), a serine/threonine protein kinase; it is also known to block mammalian target of rapamycin complex 1 (mTORC1) activity. Besides these direct effects, metformin also inhibits transcription of key gluconeogenesis genes in the liver and stimulates glucose uptake in muscle, thus lowering blood glucose and insulin levels. This decreases substrate availability and hence metabolism.[53]
Dennis et al described the role of metformin in lung cancer tumorigenesis. In this study, mice were treated with oral metformin after exposure to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). The investigators showed that metformin reduced tumor burden by up to 72%. It was also found that this effect of metformin was mediated through inhibition of mTOR in lung tissue via decreased activation of insulin-like growth factor-1 receptor/insulin receptor and Akt upstream of mTOR.[54].
Previous studies in breast cancer have examined the role of metformin in HER2/neu-expressing murine mammary adenocarcinomas that had metastasized to the lungs. Metformin treatment in the affected mice at a dose comparable to that used in humans significantly reduced the tumor size.[55]
The foregoing studies have also been supported by in vitro studies using metformin. Many recent studies have reported other effects of metformin, including reduced phosphorylation of epidermal growth factor receptor (EGFR), mitogen-activated protein kinase (MAPK), and Src. In addition, metformin lowers levels of cyclin D and E, both of which are important in the cell cycle. These effects have been studied in triple-negative breast cancer cell lines, both in vivo and in vitro, and metformin was found to reduce tumor cell proliferation and tumor growth, and to increase apoptosis. Some of these effects have also been reported to be associated with Stat3 inhibition.[56]
Implications for Dietary Recommendations in Cancer Patients
For many years, researchers have questioned the role of diet in cancer treatment. The dietary recommendation for cancer patients receiving chemotherapy, as described by the American Cancer Society, is to increase calorie and protein intake. However, it has been shown that a 20% to 40% reduction in calorie intake, or dietary restriction (DR), protects a wide variety of organisms against oxidative stress and aging.[57] This effect of fasting is mediated, in part, by a more than 50% reduction in glucose and insulin-like growth factor 1 (IGF-I) levels. Longo et al showed that treatment with starvation conditioned sensitized yeast cells (Saccharomyces cerevisiae) that expressed the oncogene-like RAS2val19 to oxidative stress, and sensitized 15 of 17 mammalian cancer cell lines to chemotherapeutic agents.[30] They also showed similar effects in human melanoma, glioma, and breast cancer cells. They found that in 4T1 breast cancer cells, this effect seemed to be mediated by increased phosphorylation of the Akt and S6 kinases, increased oxidative stress, caspase-3 cleavage, DNA damage, and apoptosis. Although calorie-restricted diets have still not been tried in clinical trials in human subjects, there is preclinical evidence to show that fasting or calorie restriction may have a role in the treatment of cancer. More studies are needed to further establish this.
Conclusion
In summary, cancer cells are dependent on glucose as the major source of energy production. The acquisition of a glycolytic phenotype by transformed cells confers a selective growth advantage to these cells and plays an important role in tumorigenesis. Interfering with aerobic glycolysis, therefore, represents a potentially effective strategy to selectively target cancer cells. As new pathways to target glucose metabolism are being discovered, it is also becoming increasingly important to identify compounds that target compensatory metabolism, which cancer cells can switch to when glycolysis has been disrupted.
Acknowledgement:This work was supported by American Cancer Society (Illinois Division) grant # 188679 to M. Shanmugam, the National American Cancer Society Research Scholar grant (RSG-11-254-01-CSM) to M. Shanmugam, and the Robert H. Lurie Comprehensive Cancer Center Director’s Fund to S. Rosen.
Financial Disclosure: The authors have no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10-29.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74.
3. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64.
4. Warburg O. On the origin of cancer cells. Science. 1956;123: 309-14.
5. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425-34.
6. Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Nat Acad Sci. 2010;107:2037-42.
7. Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51.
8. Vallejo CG, Cruz-Bermúdez A, Clemente P, et al. Evaluation of mitochondrial function and metabolic reprogramming during tumor progression in a cell model of skin carcinogenesis. Biochimie. 2013 Jan 22. [Epub ahead of print]
9. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Nat Acad Sci. 2007;104:19345-50.
10. Koppenol W H, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Rev Cancer. 2011;11:325-37.
11. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297-308.
12. Cooper GM, Hausman RE. The cell. ASM Press; Washington, DC, 2000.
13. Park HJ, Lyons JC, Ohtsubo T, Song CW. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer. 1999;80:1892.
14. Williams AC, Collard TJ, Paraskeva C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carinoma cell lines: Implications for clonal selection during colorectal carcinogenesis. Oncogene. 1999;18:3199-204.
15. Casciari JJ, Sotirchos SV, Sutherland RM. Variations in tumor cell growth rates and metabolism with oxygen concentration, glucose concentration, and extracellular pH. J Cell Physiol. 2005;151:386-94.
16. Gatenby RA, Gawlinski ET. The glycolytic phenotype in carcinogenesis and tumor invasion insights through mathematical models. Cancer Res. 2003;63:3847-54.
17. Fujiwara S, Kawano Y, Yuki H, et al. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2012;108:170-8.
18. Osthus RC, Shim H, Kim S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797-800.
19. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nature Rev Cancer. 2008;8:705-13.
20. Lu CW, Lin SC, Chen KF, et al. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283:28106-14.
21. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3.
22. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777-86.
23. Samudio I, Fiegl M, McQueen T, et al. The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008;68: 5198-205.
24. Danial NN, Gramm CF, Scorrano L, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952-6.
25. Rathmell JC, Fox CJ, Plas DR, et al. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315-28.
26. Castellani M, Carletto M, Baldini L, et al. The prognostic value of F-18 fluorodeoxyglucose bone marrow uptake in patients with recent diagnosis of multiple myeloma: a comparative study with Tc-99m sestamibi. Clin Nucl Med. 2010;35:1.
27. Zamagni E, Patriarca F, Nanni C, et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood. 2011;118:5989-95.
28. Bartel TB, Haessler J, Brown TL, et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood. 2009;114:2068-76.
29. Safdie F, Brandhorst S, Wei M, et al. Fasting enhances the response of glioma to chemo- and radiotherapy. PloS One. 2012;7:e44603.
30. Lee C, Raffaghello L, Brandhorst S, et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci Transl Med. 2012;4:124ra27.
31. Pedersen PL, Mathupala S, Rempel A, et al. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta. 2002;1555:14-20.
32. Atsumi T, Chesney J, Metz C, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62:5881-7.
33. Grassian AR, Metallo CM, Coloff JL, et al. Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Sci Signal. 2011;25:1716.
34. Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209:217-24.
35. Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3.
36. Yang W, Zheng Y, Xia Y, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature Cell Biol. 2012;14:1295-304.
37. Berruti A, Bitossi R, Gorzegno G, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. J Clin Oncol. 2002;20:4150-9.
38. Dearling JL, Qureshi U, Begent RH, Pedley RB. Combining radioimmunotherapy with antihypoxia therapy 2-deoxy-D-glucose results in reduction of therapeutic efficacy. Clin Cancer Res. 2007;13:1903-10.
39. Cao X, Bloomston M, Zhang T, et al. Synergistic antipancreatic tumor effect by simultaneously targeting hypoxic cancer cells with HSP90 inhibitor and glycolysis inhibitor. Clin Cancer Res. 2008;14:1831-9.
40. Nakashima RA, Mangan PS, Colombini M, Pedersen PL. Hexokinase receptor complex in hepatoma mitochondria: evidence from N, N’-dicyclohexlycarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry. 1986;25:1015-21.
41. Chaneton B, Hillmann P, Zheng L, et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458-62.
42. Locasale JW, Grassian AR, Melman T, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869-74.
43. Sheng SL, Liu JJ, Dai YH, et al. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBSJ. 2012;279:3898-910.
44. Adekola K, Rosen ST, Shanmugam M. Glucose transporters in cancer metabolism. Curr Opin Oncol. 2012;24:650-4.
45. Chan DA, Sutphin PD, Nguyen P, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Translat Med. 2011; 3:94ra70.
46. Murata H, Hruz PW, Mueckler M. The mechanism of insulin resistance caused by HIV protease inhibitor therapy. Sci Signal. 2000;275:20251.
47. Vyas AK, Koste, JC, Tzekov A, Hruz PW. Effects of the HIV protease inhibitor ritonavir on GLUT4 knock-out mice. J Biol Chem. 2010;285:36395-400.
48. McBrayer SK, Cheng JC, Singhal S, et al. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood. 2012;119:4686-97.
49. KovaÄeviÄ K, Morris HP. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res. 1972;32:326-33.
50. Yang C, Sudderth J, Dang T, et al. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69: 7986-93.
51. Reynolds MR, Lane AN, Robertson B, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2013 Jan 28. [Epub ahead of print]
52. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254-8.
53. Brunmair B, Staniek K, Gras F, et al. Thiazo
lidinediones, like metformin, inhibit respiratory complex IA common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052-9.
54. Memmott RM, Mercado JR, Maier CR, et al. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res. 2010;3:1066-76.
55. Anisimov VN, Egormin PA, Piskunova TS, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188-97.
56. Deng XS, Wang S, Deng A, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367-76.
57. Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharm Sci. 2010;31:89-98.

















