Molecular Pathogenesis of Cholangiocarcinoma: Implications for Disease Classification and Therapy
Khaled W. Kabbara, MD, and colleagues, research treatment options for cholangiocarcinoma.
ABSTRACT
Cholangiocarcinomas are an aggressive group of heterogeneous malignancies that affect over 210,000 individuals globally each year. Their incidence is rising, particularly in Western countries. Traditionally, cholangiocarcinomas are classified based on anatomic location of the tumor and are treated with similar cytotoxic chemotherapy despite significant molecular and genomic differences. With the rise of genetic and molecular sequencing, several driver mutations have been identified and targeted as novel therapeutic approaches. The most common genomic alterations include changes in FGFR2, IDH1, KRAS, BRAF, HER2, and the tumor suppressor p53. In addition, increased understanding of the cellular and molecular constituents of the tumor microenvironment (TME) has created opportunities for further novel therapeutic approaches. New strategies using combination therapies targeting driver mutations and various components of the TME hold promise for improved patient outcomes. This review covers the evolving molecular and therapeutic landscape of cholangiocarcinoma.
Oncology (Williston Park). 2022;36(8):492-498.
DOI: 10.46883/2022.25920971
Introduction
Cholangiocarcinomas (CCAs) comprise a rare and aggressive group of heterogeneous malignancies that combined with gallbladder cancers affect more than 210,000 individuals globally on an annual basis.1 Well-characterized risk factors include primary sclerosing cholangitis, cirrhosis, Caroli disease, viral hepatitis, cholelithiasis or choledocholithiasis, hepatolithiasis, and nonalcoholic fatty liver disease. CCA is 40 times more common in East Asia than in Western countries because of endemic infection with the liver flukes Opisthorchis viverrini, Clonorchis sinensis, and Schistosomiasis japonica as well vertical transmission of hepatitis B virus. Western countries have witnessed a 6-fold increase in incidence over the past 30 years, with some evidence suggesting a causal role for diabetes, obesity, and metabolic syndrome.2-4 Arising from malignant transformation of epithelial cholangiocytes lining biliary ducts,5 CCAs are classified anatomically by their site of origin within the biliary tree. Intrahepatic tumors arise proximal to the bifurcation of the right and left hepatic ducts. Extrahepatic tumors are divided into perihilar tumors, also known as Klatskin tumors, originating from between the bifurcation and the confluence of the cystic and hepatic ducts and distal tumors arising between the origin of the cystic duct and the ampulla of Vater. Klatskin tumors are further classified using the Bismuth-Corlette classification based on location and extension within the hilar confluence.6 Although anatomic classification has implications for locoregional therapy, approximately 80% of patients with CCA are diagnosed at an advanced stage associated with a median survival between 6 and 18 months.7 Alternative classifications based on cell of origin or growth pattern may be more relevant to disease biology but have not yet led to distinct therapeutic strategies in the advanced setting. Cytotoxic chemotherapy, based on data from clinical trials that included patients with CCA, gallbladder, and ampullary cancer, represented the only treatment option for these patients until the FDA approved pemigatinib in 2020 for patients with associated FGFR gene fusions or other gene rearrangements.8,9 The success of pemigatinib as well as the recent approval of ivosidenib for IDH1-mutated CCA represent landmark developments in the era of real-time comprehensive genomic profiling of advanced CCA for the identification of therapeutically actionable driver mutations. Furthermore, recently reported results from the phase 3 TOPAZ-1 trial (NCT03875235) revealed an overall survival (OS) benefit from the addition of the PD-L1 checkpoint inhibitor durvalumab to combination chemotherapy for patients with advanced CCA, illustrating the promise of strategies targeting intercellular interactions within the tumor microenvironment (TME).10 Thus, although anatomically classified CCAs have traditionally been grouped together as 1 entity in clinical trials, these examples highlight how increasing knowledge of the molecular basis of disease phenotypes promises a reclassification of CCA into subtypes with greater therapeutic relevance. Herein we review the most recent data associated with a molecular classification of CCA, focusing on clinically actionable targets within the cancer genome and the functional components of the TME.
Driver Mutations
The identification of mutations in oncogenes functionally relevant to the initiation and progression of CCA has led to the development of targeted therapies that are now approved for routine clinical use (Table). Next-generation sequencing platforms are commonly used to select therapy for driver mutations in CCA. In addition, companion diagnostics, including the Oncomine Dx Target Test and FoundationOne CDx, are FDA approved for drugs that target activating IDH1 mutations and FGFR fusions, respectively.11,12


TABLE. Efficacy of Targeted Therapy by Molecular Marker in Cholangiocarcinoma
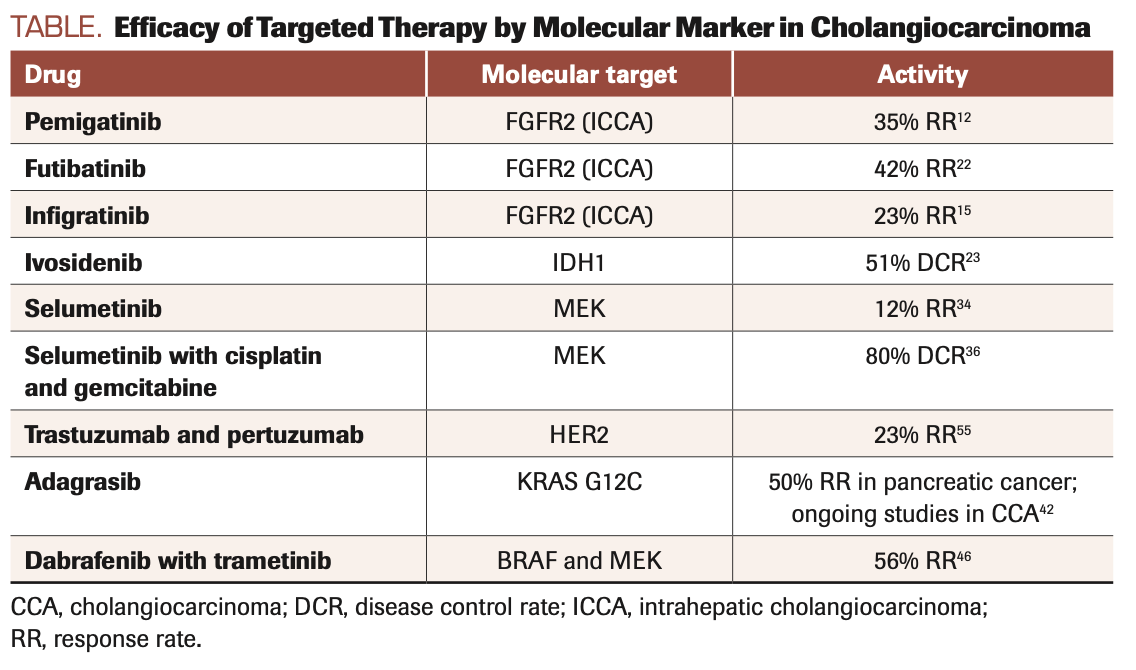
FGFR2: Four of the 5 known isoforms of FGFR function as transmembrane tyrosine kinases that exert pleiotropic effects on cell proliferation and survival in response to cognate ligand binding. FGFR mutations, amplification, and gene rearrangements including translocations and intragenic deletions have been described in a wide variety of human malignancies.13 Clonal FGFR2 gene fusions in CCA lead to ligand-independent activation of multiple signaling networks including the MAPK, PI3K-AKT, JAK-STAT, and protein kinase C pathways that in turn promote tumor progression through enhanced malignant cell proliferation, migration, and survival, as well as angiogenesis. Over 100 fusion partners of FGFR2 have been described, many unique to individual patients, and are present in 10% to 16% of intrahepatic CCAs but are rare in extrahepatic tumors.14 Pemigatinib, a selective competitive inhibitor of FGFR1/2/3, was associated with a 35% objective response rate and a median OS of 21.1 months in 107 patients with FGFR2 fusions or rearrangements enrolled in the phase 2 FIGHT-202 trial (NCT02924376), leading to FDA approval for patients with advanced CCA and FGFR2 gene fusions or rearrangements after progression on chemotherapy.15,16 Notably, none of the 20 patients in FIGHT-202 with other FGF/FGFR gene alterations achieved a response, demonstrating the specific oncogenic function of enhanced receptor dimerization resulting from gene fusion. Toxicities of pemigatinib include hyper/hypophosphatemia (12% grade 3, a class effect of FGFR inhibitors due to FGFR1 inhibition in the renal tubule) and serous retinal detachment due to subretinal fluid accumulation (4%). The role of pemigatinib in first-line therapy is being evaluated by an ongoing phase 3 clinical trial comparing the drug with gemcitabine and cisplatin chemotherapy.16,17 Infigratinib, another ATP-competitive inhibitor of FGFR1/2/3, was associated with similar results and has also been granted regulatory approval18,19; the first-line PROOF 301 trial (NCT03773302) comparing infigratinib to chemotherapy in patients with FGFR2 translocations is in progress. Multiple mutations in the kinase domain of FGFR2 confer drug resistance by interfering with the binding of competitive inhibitors. These mutations have been detected in circulating cell-free DNA and demonstrated to promote intra- and intertumoral clonal heterogeneity that evolves in parallel with resistance driven by FGFR-independent mechanisms such as loss of phosphatase and tensin homolog.20,21 The FGFR1/2/3/4 inhibitor futibatinib retains activity despite mutations that confer resistance to ATP-competitive inhibitors by binding covalently and irreversibly to the P-loop of the receptor kinase domain. Futibatinib was associated with an overall response rate of 41.7% and a median OS of 20 months in the phase 2 FOENIX-CCA2 trial (NCT02052778) and is being compared with gemcitabine and cisplatin chemotherapy in previously untreated patients with FGFR2 gene rearrangements in the ongoing phase 3 FOENIX-CCA3 trial (NCT04093362). Drugs that mitigate off-target effects by selectively inhibiting FGFR2 are in development.20-22
IDH1: IDH1 and IDH2 are metabolic enzymes that catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate and are mutated in a variety of human malignancies.23 Missense mutations in the R132 codon of IDH1 are present in 13% to 20% of intrahepatic CCAs (ICCAs) and rarely in extrahepatic CCAs (ECCAs) and perihilar CCAs, resulting in excess production of the oncometabolite R-2-hydroxyglutarate (R-2HG).3 R-2HG accumulation modifies the epigenetic state of tumor progenitor cells by altering DNA and histone methylation patterns, thereby inhibiting cellular differentiation and promoting oncogenesis.3,24 The phase 3 ClarIDHy study (NCT02989857) investigated the role of the IDH1-selective inhibitor ivosidenib in patients who had progressed on up to 2 lines of prior systemic therapy. Compared with placebo, ivosidenib significantly prolonged median progression-free survival (PFS; 2.7 vs 1.4 months; HR, 0.37; P <.0001) and OS adjusted for crossover (10.8 vs 5.1 months; HR, 0.49; P <.001).25-27 The percentage of patients treated with ivosidenib who were progression free at 6 and 12 months was 32% and 22%, respectively, whereas no patients in the placebo arm remained progression free at 6 months. Ivosidenib is administered orally and is well tolerated; low-grade nausea, diarrhea, and fatigue were the most common treatment-emergent adverse events, and there were low rates of drug discontinuation and dose reduction. Ivosidenib is FDA approved for patients with previously treated locally advanced or metastatic CCA with an IDH1 mutation. Resistance-promoting receptor tyrosine kinase mutations as well as secondary IDH1 mutations that inhibit drug binding and restore cellular R-2HG levels have been described in acute myeloid leukemia, but mechanisms of ivosidenib resistance in CCA have yet to be elucidated.28
MAPK/KRAS: The MAPK pathway includes several intermediaries that play a central role in carcinogenesis, and mutated forms are common drivers of CCA.29 These protein kinases, which include Ras, Raf, MEK, and ERK, are involved in signal transduction pathways that modulate a variety of processes that impact cellular pathophysiology. Mutations of KRAS and BRAF that result in constitutive activation of the protein kinase are particularly common.3,30 RAS mutations, particularly in KRAS, are found in 38% of ECCAs,31 but in less than 10% of ICCAs. These activating mutations are most commonly found in exon 2 and less commonly in exons 3 and 4.32
Differences in the genomic makeup between ICCA and ECCA are likely partially related to differences in the cell of origin. Periductal glandular epithelial cells surrounding larger bile ducts, including the common bile duct, may constitute the more common cell of origin for ECCA. These cells are associated with distinct molecular alterations, specifically RAS mutations, which are linked to lower PFS and OS.33-37 The high frequency of RAS mutations in ECCA may be related to the poor response to chemotherapy that characterizes ECCA, with outcomes similar to RAS-mutated pancreatic adenocarcinoma.
Targeting the MAPK pathway in CCA has not yet led to consistent therapeutic benefit. Strategies that target MEK, an intermediary downstream of Ras, have been investigated. Bekaii-Saab et al reported a 12% response rate with selumetinib monotherapy among 28 patients with metastatic biliary cancers (not selected for specific alterations), but to our knowledge no other studies have since shown high response rates with this strategy.38 Selumetinib in combination with gemcitabine and cisplatin chemotherapy was associated with a disease control rate of 80% and median OS of 9.8 months.29,39,40 Although pan-KRAS inhibitors have been elusive, mutation-specific KRAS G12C inhibitors have shown high response rates in KRAS G12C–mutated lung cancer.41,42 KRAS G12C mutations comprise up to 7.1% of KRAS mutations in CCA.43 Although CCAs have not been heavily represented in KRAS G12C inhibitor clinical trials, at least 1 patient had stable disease with sotorasib in the CodeBreaK 100 trial (NCT03600883).44 Adagrasib, an irreversible covalent inhibitor that binds to KRAS G12C, is now being investigated, and a partial remission was reported in a patient with CCA in the KRYSTAL-1 trial (NCT03785249).45,46 Other inhibitors of specific KRAS mutations are now in development, including inhibitors of KRAS G12D.47
BRAF: BRAF mutations occur in approximately 5% of CCA and are mutually exclusive from KRAS.29 They have been more commonly described in ICCAs.48 BRAF is downstream of KRAS, with the most common mutation at V600E, resulting in strong activation of RAF kinase and RAS-independent signaling. Other less common mutations include class 2 mutations, which result in intermediate RAF kinase activation, and the “kinase-dead” class 3 mutations that result in BRAF activation through a negative feedback loop.3 Extrapolating from treatment trials in cancer subsets where BRAF V600 mutations are more common, such as melanoma and colorectal cancer, has led to the investigation of MEK and BRAF inhibition in this subset of CCA. The phase 2 ROAR trial (NCT02034110), for example, demonstrated that combination therapy with dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor)49 resulted in a response rate of 56% and a median OS of 15 months.50 Several basket trials, including TAPUR (NCT02693535) and NCI-Match (NCT02465060), are also investigating combination therapy in BRAF-mutated CCA.51,52
HER2: HER2, encoded by the ERBB2 gene, is a member of the human epidermal growth factor receptor family and is a plasma membrane-bound receptor tyrosine kinase.53 It interacts with multiple signaling nodules and initiates multiple different signaling pathways. Targeting of this pathway has changed the landscape of breast, gastric, and colorectal cancers.54,55 Ligand binding causes dimerization of the HER receptor, leading to activation of the tyrosine kinase and downstream signaling cascades including the MAPK pathway, which in turn results in enhanced cellular proliferation.56,57 Deregulation of these receptors plays a role in tumorigenesis. In a meta-analysis, HER2 overexpression was seen in 20% of patients with ECCA and is rarely found in ICCA.57
The combination of trastuzumab and pertuzumab resulted in a response rate of 23% in HER2-amplified or -overexpressed metastatic biliary tract cancer previously treated, which included CCA.58 The median duration of response was 10.8 months, and the median OS was 10.9 months. Several other investigations of HER2-targeted agents in CCA are ongoing in basket trials, as well as a Korean study of trastuzumab combined with folinic acid, fluorouracil, and oxaliplatin, or FOLFOX, in pretreated HER2-amplified metastatic biliary tract cancer.59,60
p53:TP53 is a tumor suppressor gene that is also present in different types of CCA.29 Lowery et al observed TP53 mutations, common in multiple biliary tract malignancies and pancreatic cancer, in 49% of ECCAs and less than 20% of ICCAs.31 TP53 mutations are seen at a higher prevalence in fluke-related CCA and hepatitis B antigen seropositive patients.29,61-63 Strategies targeting p53 include degradation of mutant p53, restoration of wild-type p53 through epigenetic modification and clustered regularly interspaced short palindromic repeats technology, and immunotherapy targeting cells expressing mutant TP53.64 Although these strategies are currently under various stages of investigation, their potential application to patients with CCA remains uncertain.64
The Tumor Microenvironment
The TME consists of heterogeneous cell types including tumor-infiltrating lymphocytes (TILs) and natural killer cells; cancer-associated fibroblasts (CAFs); tumor-associated macrophages (TAMs) and myeloid cells; endothelial cells and pericytes; and a desmoplastic extracellular matrix (ECM) consisting of proteoglycans and soluble factors. A variety of treatment strategies directed against various constituents of the TME are being explored, and here we highlight efforts to target TILs, CAFs, TAMs, and the ECM.23,65
TILs: Interactions between malignant cells and TILs promote tumor progression in part through the activation of immune checkpoints including PD-1 (and its ligand PD-L1) and CTLA-4 that result in exhaustion of cytotoxic CD8+ lymphocytes and upregulation of CD4+CD25+FOXP3+ regulatory T cells.66 The aforementioned global phase 3 TOPAZ-1 trial established the efficacy of combining the PD-L1 checkpoint inhibitor durvalumab with chemotherapy in previously untreated patients, improving survival at 2 years from 10.4% with chemotherapy alone to 24.9% with combined therapy. TOPAZ-1 enrolled 685 patients with ICCA (56%), ECCA (19%), and gallbladder cancer (25%), and benefit was reported in all subtypes.10
Dual checkpoint therapy with the PD-1 antibody nivolumab and the CTLA-4 antibody ipilimumab has also demonstrated early efficacy in this disease type, but with differential responses depending on anatomic location of the tumor, with associated objective responses in 5 of 16 patients with advanced ICCA but 0 of 10 patients with ECCA.67 The combination of nivolumab and ipilimumab is now being compared with nivolumab, gemcitabine, and cisplatin in a multicenter, randomized phase 2 study of previously untreated patients with advanced CCA.68
PD-L1, microsatellite instability (MSI), and high tumor mutational burden (TMB) have all been explored as biomarkers for selecting patients for immunotherapy.69-71 A phase 2 trial reported PD-L1 expression in 43% of CCAs, and consistent with cumulative experience across a broad variety of malignancies, TOPAZ-1 reported greater relative benefit in patients with PD-L1 expression with a hazard ratio for OS of 0.79 in 58% of patients with PD-L1 tumor area positivity (TAP) of 1% or greater vs 0.86 in 30% of patients with PD-L1 TAP of less than 1%.10 However, given the observation of responses even in patients without protein expression, PD-L1 immunohistochemistry is an imperfect biomarker for patient selection. MSI and high TMB are postulated to predict responses to checkpoint inhibitor therapy by increasing neoantigen expression and immune activation.72 Both are FDA-approved biomarkers for selecting patients for the PD-1 antibody pembrolizumab agnostic of tumor type. MSI (or its correlate, deficient DNA mismatch repair protein expression) has been reported in 1% to 10% of CCAs, with greater prevalence in ICCAs vs ECCAs.67,70 The KEYNOTE-158 trial (NCT02628067) included 22 patients with CCA, of whom 9 (41%) had an objective response to pembrolizumab.73 High TMB is seen in less than 5% of CCAs, but there is scant evidence that it predicts response to checkpoint inhibitors.74 Genomic approaches combining laser-capture microdissection of tumor stroma with functionally validated computational annotation of TME components may offer improved disease classification and identification of an immunogenic subtype sensitive to checkpoint inhibitor therapy.70 Moreover, strategies targeting immune checkpoints beyond PD-1/PD-L1 including LAG3, TIM-3, TIGIT, and BTLA as well as costimulatory receptors including OX40, 4-1BB, GITR, and ICOS remain under investigation across a broad variety of malignancies and may provide additional benefit as well as markers for improved patient selection.69,70


FIGURE. Therapeutic Strategies for Targeting the Cholangiocarcinoma Microenvironment
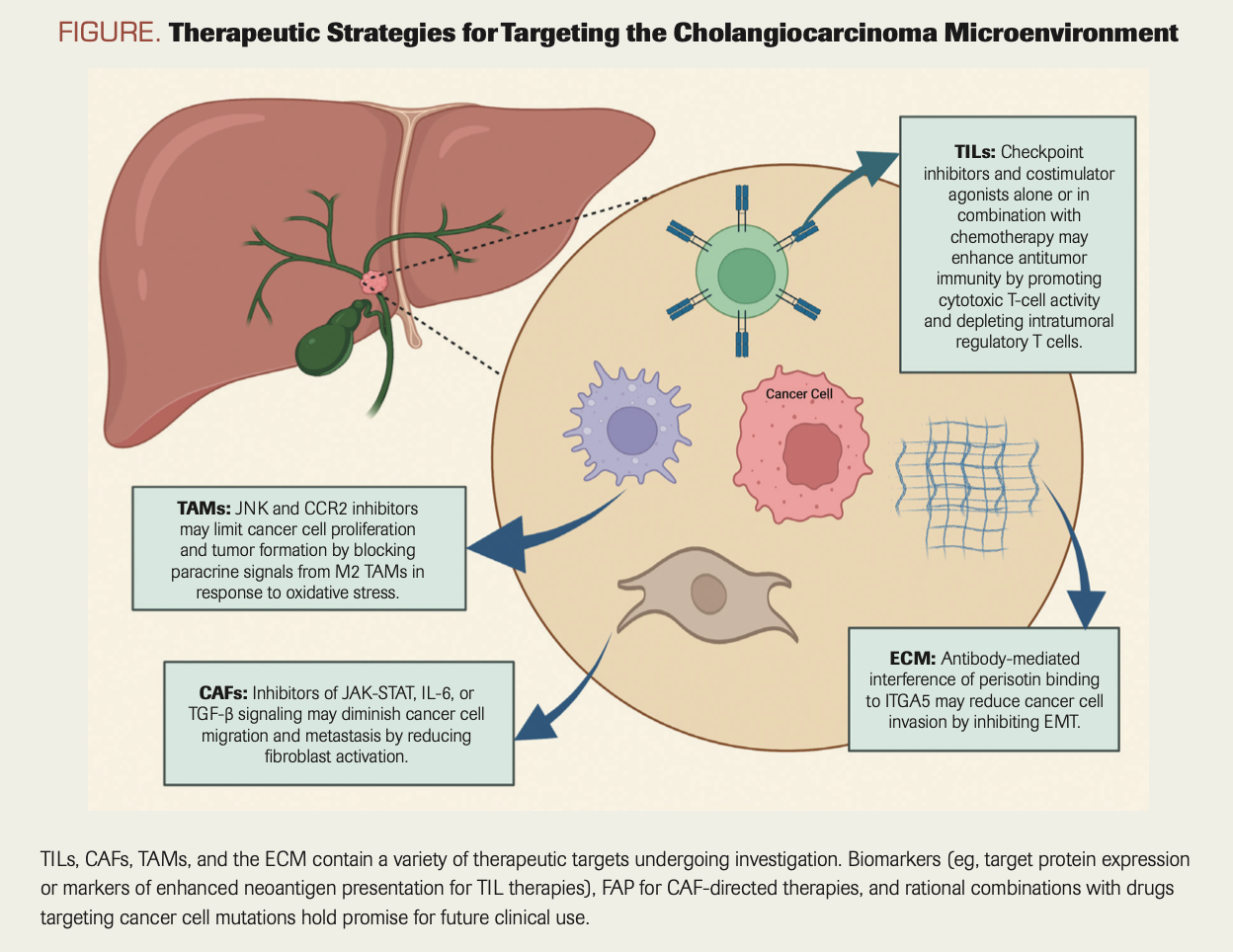
CAFs: Comprising the majority of cells within the tumor stroma, CAFs in CCAs are thought to arise from a variety of normal precursors including hepatic stellate cells, portal fibroblasts, and bone marrow–derived mesenchymal cells.75,76 Far from representing a passive reaction of normal host tissue to malignant tumor development, the pleiotropic mechanisms whereby CAFs actively enhance cancer cell migration, metastasis, and chemoresistance have been elucidated using a variety of model systems across a broad range of solid tumors and present opportunities for therapeutic intervention.75-81 For example, inflammatory cytokines within the TME including IL-6 and TGF-β promote stromal remodeling and cancer cell migration through JAK-STAT–dependent activation of actomyosin contractility within CAFs.82-85 IL-6 antibodies and JAK inhibitors that have been FDA approved for autoimmune and myeloproliferative diseases are in clinical trials for solid tumors including CCA, and novel small molecule inhibitors of JAK-STAT signaling including S63845 and AZD1480 are being investigated in preclinical studies.85,86 Strategies targeting TGF-β have been complicated by incomplete understanding of its distinct tumor-suppressive and tumor-promoting functions mediated through canonical SMAD and non-SMAD signaling cascades. The bifunctional fusion protein bintrafusp-α, composed of the extracellular domain of the TGF-β receptor TβRII and human IgG1 antibody targeting PD-L1, demonstrated an objective response rate of 10.1% in patients previously treated with platinum chemotherapy, but a first-line trial evaluating the drug in combination with chemotherapy was discontinued due to futility.87,88 Additional strategies targeting CAFs using antibodies or CAR T cells directed against the serine protease fibroblast activation protein as well as other CAF markers hold promise but remain in need of further development, including the identification of biomarker-defined subsets with a higher likelihood of benefit.89
TAMS: TAMs are derived from resident hepatic macrophages known as Kupffer cells and from circulating CD14+/CD16+ monocytes that infiltrate the TME in response to chemoattractant proteins including MCP/CCL2 and CSF1.76,90-93 Similar to CAFs, TAMs synthesize stromal remodeling enzymes including matrix metalloproteinases to create a suitable environment for tumor growth and cancer cell migration.94 In addition, TAMs play a dual role in modulating the immune response through CCR2/CCL2-mediated polarization into M1 and M2 subsets promoting inflammation and immunosuppression, respectively.95,96 M2 TAMs inhibit cytotoxic T cells and facilitate angiogenesis by expressing PD-L1, CTLA-4, and VEGF, fostering an immunosuppressive milieu conducive to tumor growth and metastasis.71,80,81,97,98
Preclinical studies have investigated strategies for reducing TAM infiltration, M2 TAM polarization, and cooperative signals between TAMs and malignant cells.96,99 For example, mouse models of ICCA demonstrate that Kupffer cells produce tumor necrosis factor (TNF) in response to oxidative stress within the liver, and that TNF in turn leads to paracrine activation of JNK-mediated cholangiocellular overgrowth and tumor formation.100 Small molecule inhibition of JNK abrogated tumor growth in mice and the proliferation of human ICCA cell lines. Further supported by the observation that JNK activation has been reported in 80% of human ICCAs, targeting TNF-JNK signaling between TAMs and malignant cells with JNK inhibitors is of interest for clinical development.100,101
In addition, early clinical efforts in pancreatic cancer targeting TAMs by inhibiting CCR2 in combination with cytotoxic chemotherapy may hold similar promise for patients with CCA.102 However, a study using a genetically engineered mouse model of CCA demonstrated that inhibition of TAM CCR2 led to a compensatory increase in granulocytic myeloid-derived suppressor cells and that dual inhibition of both cell types in combination with immune checkpoint blockade was required to potentiate tumor regression.71 Clinical strategies that leverage a more sophisticated understanding of the immune response to CCAs will require careful patient selection based on personalized characterization of the TME.
ECM: ECM components, including fibrillar proteins, glycoproteins, and proteoglycans, contribute to tumor formation and progression through a variety of mechanisms that suggest potential therapeutic targets.103 For example, the CAF-secreted matricellular protein periostin contributes to cell migration and metastasis in CCAs by promoting malignant cell epithelial-to-mesenchymal transition (EMT).104 Antibody-mediated blockade or inhibition of the periostin receptor integrin alpha 5 on CCA cells reduced CCA proliferation and invasion by downregulating the EMT-promoting transcription factor TWIST2, and higher expression levels of TWIST2 in human CCA samples are associated with poor prognosis.104,105 Nonetheless, ECM-targeting strategies in patients based on promising preclinical data have largely fallen short in the clinical setting, as exemplified by the negative phase 3 trial in patients with pancreatic cancer evaluating the addition of recombinant human hyaluronidase PEGPH20 to gemcitabine and nanoparticle albumin-bound paclitaxel.106 Thus, indiscriminate efforts to target the ECM in CCA similarly may fail without improved patient selection via enhanced mechanistic understanding of ECM components in different disease subsets and stages of tumor development.
Conclusion
To date, CCA has generally been treated as a single entity in clinical trials. Treatment paradigms have not significantly varied based on location in the biliary tree. However, the advancement of molecular diagnostics including genomic sequencing has shed new light on the unique subtypes within the broad classification of CCA. Indeed, extrahepatic and intrahepatic CCA differ so greatly in molecular findings that it seems reasonable to consider them as separate diseases, for which unique clinical trials should be performed. Intrahepatic CCA has been associated with longer OS per stage and often has clear driver mutations, such as FGFR fusions or IDH1 mutations, whereas extrahepatic and hilar CCA share genomic similarities with pancreatic adenocarcinoma.
Molecular profiling has been a cornerstone for new therapeutic endeavors and personalized medicine. In addition, increased understanding of all
components of the TME and their pleiotropic effects on tumor progression will allow for new avenues of therapeutic discovery. Combination therapy that targets the tumor stroma in conjunction with actionable mutations represents a promising strategy, which in turn may enhance clinically relevant molecular classification and patient selection. For example, the attenuating effect of pharmacologic IDH1 inhibition on ICCA in a genetically engineered mouse model was limited by immune checkpoint activation and recruitment of regulatory T cells; combination therapy with an anti–CTLA-4 antibody and IDH1 inhibitor led to synergistic antitumor effects meriting further investigation.107
Rapidly expanding technology for characterizing circulating tumor DNA (ctDNA) and metabolites using liquid biopsies may circumvent the need for tumor sequencing, although the utility of ctDNA in the clinic may be limited by the absence of spatial resolution for characterizing the outgrowth of resistant clones.108 Moreover, ctDNA alone will not permit real-time analysis and tailored targeting of the TME. Investigational molecular imaging using novel tracers coupled to radioisotopes, bioluminescent probes, and fluorescent reporters holds promise for improved diagnosis and in vivo target validation in early drug development trials.109 Finally, ongoing efforts to characterize the role of host genetic background through genome-wide association studies of cancer predisposition as well as the gut microbiome may further aid in the molecular classification and treatment of patients with CCA.39,110
Disclosure: The authors have no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
Authors’ affiliation:
Khaled W. Kabbara, MD; timothy Cannon, MD; Arthur Winer, MD; and Raymond Wadlow, MD
INOVA Schar Cancer Institute
Corresponding Author:
Address: Inova Schar Cancer Institute, 8081 Innovation Park Dr, Fairfax, VA 22031
Tel: 571-472-1180
Fax: 571-472-1197
Email: raymond.wadlow@inova.org
References
- Ouyang G, Liu Q, Wu Y, et al. The global, regional, and national burden of gallbladder and biliary tract cancer and its attributable risk factors in 195 countries and territories, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study 2017. Cancer. 2021;127(13):2238-2250. doi:10.1002/cncr.33476
- Patel T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology. 2001;33(6):1353-1357. doi:10.1053/jhep.2001.25087
- Valle JW, Kelley RK, Nervi B, Oh DY, Zhu AX. Biliary tract cancer. Lancet. 2021;397(10272):428-444. doi:10.1016/S0140-6736(21)00153-7
- Shaib YH, Davila JA, McGlynn K, El-Serag HB. Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol. 2004;40(3):472-477. doi:10.1016/j.jhep.2003.11.030
- Khan AS, Dageforde LA. Cholangiocarcinoma. Surg Clin North Am. 2019;99(2):315-335. doi:10.1016/j.suc.2018.12.004
- Suarez-Munoz MA, Fernandez-Aguilar JL, Sanchez-Perez B, et al. Risk factors and classifications of hilar cholangiocarcinoma. World J Gastrointest Oncol. 2013;5(7):132-138. doi:10.4251/wjgo.v5.i7.132
- Bridgewater J, Lopes A, Palmer D, et al. Quality of life, long-term survivors and long-term outcome from the ABC-02 study. Br J Cancer. 2016;114(9):965-971. doi:10.1038/bjc.2016.64
- Khan SA, Tavolari S, Brandi G. Cholangiocarcinoma: epidemiology and risk factors. Liver Int. 2019;39(suppl 1):19-31. doi:10.1111/liv.14095
- FDA grants accelerated approval to pemigatinib for cholangiocarcinoma with an FGFR2 rearrangement or fusion. News release. FDA. April 17, 2020. Accessed July 19, 2022. https://bit.ly/3IQ9VDd
- Oh DY, He AR, Qin S, et al. A phase 3 randomized, double-blind, placebo-controlled study of durvalumab in combination with gemcitabine plus cisplatin (GemCis) in patients (pts) with advanced biliary tract cancer (BTC): TOPAZ-1. J Clin Oncol. 2022;40(suppl 4):378-378. doi:10.1200/JCO.2022.40.4_suppl.37811
- FDA Approves NGS-Based Companion Diagnostic for Previously Treated IDH1-Mutated Cholangiocarcinoma. News release. Thermo Fisher Scientific. August 25, 2021. Accessed July 19, 2022. https://prn.to/3aN949U
- Recently-approved devices. FoundationOne CDx - P170019/S013. US Food and Drug Administration. Updated May 12, 2020. Accessed June 17, 2022. https://bit.ly/3Pv5rUE
- Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318-332. doi:10.1038/nrc.2017.8
- De Luca A, Esposito Abate R, Rachiglio AM, et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21(18):6856. doi:10.3390/ijms21186856
- Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671-684. doi:10.1016/S1470-2045(20)30109-1
- Weaver A, Bossaer JB. Fibroblast growth factor receptor (FGFR) inhibitors: a review of a novel therapeutic class. J Oncol Pharm Pract. 2021;27(3):702-710. doi:10.1177/1078155220983425
- A study to evaluate the efficacy and safety of pemigatinib versus chemotherapy in unresectable or metastatic cholangiocarcinoma (FIGHT-302). ClinicalTrials.gov. Upated June 9, 2022. Accessed March 8, 2022. https://bit.ly/3uVyLfw
- Fala L. Truseltiq (infigratinib) new targeted therapy FDA approved for advanced or metastatic cholangiocarcinoma harboring FGFR2 alterations.J Hematol Oncol Pharm. 2021;11(4). https://bit.ly/3PlZqdp
- Salati M, Caputo F, Baldessari C, et al. The evolving role of FGFR2 inhibitors in intrahepatic cholangiocarcinoma: from molecular biology to clinical targeting. Cancer Manag Res. 2021;13:7747-7757. doi:10.2147/CMAR.S330710
- Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion–positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252-263. doi:10.1158/2159-8290.CD-16-1000
- Goyal L, Borad M, Subbiah V, et al. First results of RLY-4008, a potent and highly selective FGFR2 inhibitor in a first-in-human study in patients with FGFR2-altered cholangiocarcinoma and multiple solid tumors. Mol Cancer Ther. 2021;20(suppl 12):P02-02. doi:10.1158/1535-7163.targ-21-p02-02
- Borad MJ, Bridgewater JA, Morizane C, et al. A phase III study of futibatinib (TAS-120) versus gemcitabine-cisplatin (gem-cis) chemotherapy as first-line (1L) treatment for patients (pts) with advanced (adv) cholangiocarcinoma (CCA) harboring fibroblast growth factor receptor 2 (FGFR2) gene rearrangements (FOENIX-CCA3). J Clin Oncol. 2020;38(suppl 4):TPS600. doi:10.1200/JCO.2020.38.4_suppl.TPS600
- Xiang X, Liu Z, Zhang C, et al. IDH mutation subgroup status associates with intratumor heterogeneity and the tumor microenvironment in intrahepatic cholangiocarcinoma. Adv Sci (Weinh). 2021;8(17):e2101230. doi:10.1002/advs.202101230
- Boscoe AN, Rolland C, Kelley RK. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: a systematic literature review. J Gastrointest Oncol. 2019;10(4):751-765. doi:10.21037/jgo.2019.03.10
- Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: ready for "prime time" in biliary tract cancer. J Hepatol. 2020;73(1):170-185. doi:10.1016/j.jhep.2020.03.007
- Li Y, Song Y, Liu S. The new insight of treatment in cholangiocarcinoma. J Cancer. 2022;13(2):450-464. doi:10.7150/jca.68264
- Zhu AX, Macarulla T, Javle MM, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical ClarIDHy trial. JAMA Oncol. 2021;7(11):1669-1677. doi:10.1001/jamaoncol.2021.3836
- Choe S, Wang H, DiNardo CD, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020;4(9):1894-1905. doi:10.1182/bloodadvances.2020001503
- Sohal DP, Shrotriya S, Abazeed M, Cruise M, Khorana A. Molecular characteristics of biliary tract cancer. Crit Rev Oncol Hematol. 2016;107:111-118. doi:10.1016/j.critrevonc.2016.08.013
- De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12(6):594-603. doi:10.1016/S1470-2045(10)70209-6
- Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res. 2018;24(17):4154-4161. doi:10.1158/1078-0432.CCR-18-0078
- Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003-1010. doi:10.1038/ng.3375
- Komuta M, Govaere O, Vandecaveye V, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology. 2012;55(6):1876-1888. doi:10.1002/hep.25595
- Liau JY, Tsai JH, Yuan RH, Chang CN, Lee HJ, Jeng YM. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod Pathol. 2014;27(8):1163-1173. doi:10.1038/modpathol.2013.241
- Komuta M, Spee B, Vander Borght S, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47(5):1544-1556. doi:10.1002/hep.22238
- Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557-588. doi:10.1038/s41575-020-0310-z
- Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9(12):e115383. doi:10.1371/journal.pone.0115383
- Bekaii-Saab T, Phelps MA, Li X, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29(17):2357-2363. doi:10.1200/JCO.2010.33.9473
- Chang YC, Chen MH, Yeh CN, Hsiao M. Omics-based platforms: current status and potential use for cholangiocarcinoma. Biomolecules. 2020;10(10):1377. doi:10.3390/biom10101377
- Bridgewater J, Lopes A, Beare S, et al. A phase 1b study of selumetinib in combination with cisplatin and gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer. 2016;16:153. doi:10.1186/s12885-016-2174-8
- Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384(25):2371-2381. doi:10.1056/NEJMoa2103695
- Zheng X, Luo J, Liu W, Ashby CR Jr, Chen ZS, Lin L. Sotorasib: a treatment for non-small cell lung cancer with the KRAS G12C mutation. Drugs Today (Barc). 2022;58(4):175-185. doi:10.1358/dot.2022.58.4.3400573
- Zhou SL, Xin HY, Sun RQ, et al. Association of KRAS variant subtypes with survival and recurrence in patients with surgically treated intrahepatic cholangiocarcinoma. JAMA Surg. 2022;157(1):59-65. doi:10.1001/jamasurg.2021.5679
- Strickler JH, Fakih M, Price TJ, et al. 83MO AMG 510, a novel small molecule inhibitor of KRAS (G12C), for patients (pts) with advanced gastrointestinal (GI) cancers: results from the CodeBreaK100 phase I trial. Ann Oncol. 2020;31(suppl 6):S1274-S1275. doi:10.1016/j.annonc.2020.10.103
- Janne PA, Rybkin II, Spira A, et al. KRYSTAL-1: updated safety and efficacy data with adagrasib (MRTX849) in NSCLC with KRASG12C mutation from a phase 1/2 study. Eur J Cancer. 2020;138(suppl 2):S1-S2. doi:10.1016/S0959-8049(20)31076-5
- Bekaii-Saab TS, Spira AI, Yaeger R, et al. KRYSTAL-1: Updated activity and safety of adagrasib (MRTX849) in patients (Pts) with unresectable or metastatic pancreatic cancer (PDAC) and other gastrointestinal (GI) tumors harboring a KRASG12C mutation. J Clin Oncol. 2022;40(suppl 4):519-519. doi:10.1200/jco.2022.40.4_suppl.519
- Mao Z, Xiao H, Shen P, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8(1):5. doi:10.1038/s41421-021-00368-w
- Zhu AX, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol. 2014;21(12):3827-3834. doi:10.1245/s10434-014-3828-x
- Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020;21(9):1234-1243. doi:10.1016/S1470-2045(20)30321-1
- Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol. 2022;33(4):406-415. doi:10.1016/j.annonc.2021.12.014
- Targeted therapy directed by genetic testing in treating patients with advanced refractory solid tumors, lymphomas, or multiple myeloma (the MATCH Screening Trial). ClinicalTrials.gov. Updated July 15, 2022. Accessed April 24, 2022. https://bit.ly/3yJ1pBs
- TAPUR: testing the use of food and drug administration (FDA) approved drugs that target a specific abnormality in a tumor gene in people with advanced stage cancer (TAPUR). ClinicalTrials.gov. Updated June 1, 2022. Accessed April 24, 2022. https://bit.ly/3zeMr7I
- Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051-2058. doi:10.1172/JCI32278
- Sartore-Bianchi A, Trusolino L, Martino C, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738-746. doi:10.1016/S1470-2045(16)00150-9
- Meric-Bernstam F, Hurwitz H, Raghav KPS, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20(4):518-530. doi:10.1016/S1470-2045(18)30904-5
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39-51. doi:10.1056/NEJMra043186
- Galdy S, Lamarca A, McNamara MG, et al. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: a potential therapeutic target? Cancer Metastasis Rev. 2017;36(1):141-157. doi:10.1007/s10555-016-9645-x
- Javle M, Borad MJ, Azad NS, et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021;22(9):1290-1300. doi:10.1016/S1470-2045(21)00336-3
- Trastuzumab-pkrb combined with modified FOLFOX-6 in biliary tract cancer patients progressed on first line therapy. ClinicalTrials.gov. Updated January 25, 2021. Accessed April 24, 2022. https://bit.ly/3PlZycT
- Rizzo A, Ricci AD, Bonucci C, et al. Experimental HER2- targeted therapies for biliary tract cancer. Expert Opin Investig Drugs. 2021;30(4):389-399. doi:10.1080/13543784.2021.1854724
- Chan-On W, Nairismägi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45(12):1474-1478. doi:10.1038/ng.2806
- Saleh M, Virarkar M, Bura V, et al. Intrahepatic cholangiocarcinoma: pathogenesis, current staging, and radiological findings. Abdom Radiol (NY). 2020;45(11):3662-3680. doi:10.1007/s00261-020-02559-7
- Wu CE, Pan YR, Yeh CN, Lunec J. Targeting p53 as a future strategy to overcome gemcitabine resistance in biliary tract cancers. Biomolecules. 2020;10(11):1474. doi:10.3390/biom10111474
- Zhu G, Pan C, Bei JX, et al. Mutant p53 in cancer progression and targeted therapies. Front Oncol. 2020;10:595187. Published online November 6, 2020. doi:10.3389/fonc.2020.595187
- Zhang M, Yang H, Wan L, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. 2020;73(5):1118-1130. doi:10.1016/j.jhep.2020.05.039
- Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298-306. doi:10.1038/nrc3245
- Klein O, Kee D, Nagrial A, et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: subgroup analysis of a phase 2 nonrandomized clinical trial. JAMA Oncol. 2020;6(9):1405-1409. doi:10.1001/jamaoncol.2020.2814
- Study of nivolumab in combination with gemcitabine/cisplatin or ipilimumab for patients with advanced unresectable biliary tract cancer. ClinicalTrials.gov. Updated February 1, 2022. Accessed April 18, 2022. https://bit.ly/3yULD6O
- Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. 2022;19(1):37-50. doi:10.1038/s41571-021-00552-7
- Job S, Rapoud D, Dos Santos A, et al. Identification of four immune subtypes characterized by distinct composition and functions of tumor microenvironment in intrahepatic cholangiocarcinoma. Hepatology. 2020;72(3):965-981. doi:10.1002/hep.31092
- Loeuillard E, Yang J, Buckarma E, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest. 2020;130(10):5380-5396. doi:10.1172/JCI137110
- Zhang R, Li Q, Fu J, et al. Comprehensive analysis of genomic mutation signature and tumor mutation burden for prognosis of intrahepatic cholangiocarcinoma. BMC Cancer. 2021;21(1):112. doi:10.1186/s12885-021-07788-7
- Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38(1):1-10. doi:10.1200/JCO.19.02105
- Klempner SJ, Fabrizio D, Bane S, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25(1):e147-e159. doi:10.1634/theoncologist.2019-0244
- Affo S, Yu LX, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol. 2017;12:153-186. doi:10.1146/annurev-pathol-052016-100322
- Fabris L, Perugorria MJ, Mertens J, et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019;39(suppl 1):63-78. doi:10.1111/liv.14098
- Okabe H, Beppu T, Hayashi H, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. 2009;16(9):2555-2564. doi:10.1245/s10434-009-0568-4
- Dranoff JA, Wells RG. Portal fibroblasts: underappreciated mediators of biliary fibrosis. Hepatology. 2010;51(4):1438-1444. doi:10.1002/hep.23405
- Russo FP, Alison MR, Bigger BW, et al. The bone marrow functionally contributes to liver fibrosis. Gastroenterology. 2006;130(6):1807-1821. doi:10.1053/j.gastro.2006.01.036
- Fabris L, Sato K, Alpini G, Strazzabosco M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology. 2021;73(suppl 1):75-85. doi:10.1002/hep.31410
- Louis C, Edeline J, Coulouarn C. Targeting the tumor microenvironment in cholangiocarcinoma: implications for therapy. Expert Opin Ther Targets. 2021;25(2):153-162. doi:10.1080/14728222.2021.1882998
- Clapéron A, Mergey M, Aoudjehane L, et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology. 2013;58(6):2001-2011. Published correction appears in Hepatology. 2014;60(2):770. doi:10.1002/hep.26585
- Albrengues J, Bertero T, Grasset E, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204. doi:10.1038/ncomms10204
- Sanz-Moreno V, Gaggioli C, Yeo M, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20(2):229-245. doi:10.1016/j.ccr.2011.06.018
- Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99-115. doi:10.1038/s41573-018-0004-1
- Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16(6):487-497. doi:10.1016/j.ccr.2009.10.015
- Yoo C, Oh DY, Choi HJ, et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with pretreated biliary tract cancer. J Immunother Cancer. 2020;8(1):e000564. doi:10.1136/jitc-2020-000564
- Gemcitabine plus cisplatin with or without bintrafusp alfa (M7824) in participants with 1L biliary tract cancer (BTC). ClinicalTrials.gov. Updated June 13, 2022. Accessed April 24, 2022. https://bit.ly/3uX1gsX
- Bughda R, Dimou P, D'Souza RR, Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther. 2021;10:313-323. doi:10.2147/ITT.S291767
- Subimerb C, Pinlaor S, Khuntikeo N, et al. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol Med Rep. 2010;3(4):597-605. doi:10.3892/mmr_00000303
- Høgdall D, Lewinska M, Andersen JB. Desmoplastic tumor microenvironment and immunotherapy in cholangiocarcinoma. Trends Cancer. 2018;4(3):239-255. doi:10.1016/j.trecan.2018.01.007
- Kitano Y, Okabe H, Yamashita YI, et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br J Cancer. 2018;118(2):171-180. doi:10.1038/bjc.2017.401
- Goeppert B, Frauenschuh L, Zucknick M, et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br J Cancer. 2013;109(10):2665-2674. doi:10.1038/bjc.2013.610
- Raggi C, Correnti M, Sica A, et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J Hepatol. 2017;66(1):102-115. doi:10.1016/j.jhep.2016.08.012
- Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy Immunity. 2014;41(1):49-61. Published correction appears in Immunity. 2014;41(5):866. doi:10.1016/j.immuni.2014.06.010
- Ge Z, Ding S. The crosstalk between tumor-associated macrophages (TAMs) and tumor cells and the corresponding targeted therapy. Front Oncol. 2020;10:590941. Published online November 3, 2020. doi:10.3389/fonc.2020.590941
- Lim YJ, Koh J, Kim K, et al. Clinical implications of cytotoxic T lymphocyte antigen-4 expression on tumor cells and tumor-infiltrating lymphocytes in extrahepatic bile duct cancer patients undergoing surgery plus adjuvant chemoradiotherapy. Target Oncol. 2017;12(2):211-218. doi:10.1007/s11523-016-0474-1
- Kitano Y, Yamashita YI, Nakao Y, et al. Clinical significance of PD-L1 expression in both cancer and stroma cells of cholangiocarcinoma patients. Ann Surg Oncol. 2020;27(2):599-607. doi:10.1245/s10434-019-07701-4
- Zhou M, Wang C, Lu S, et al. Tumor-associated macrophages in cholangiocarcinoma: complex interplay and potential therapeutic target. EBioMedicine. 2021;67:103375. doi:10.1016/j.ebiom.2021.103375
- Yuan D, Huang S, Berger E, et al. Kupffer cell-derived Tnf triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell. 2017;31(6):771-789.e6. doi:10.1016/j.ccell.2017.05.006
- Chen C, Nelson LJ, Ávila MA, Cubero FJ. Mitogen-activated protein kinases (MAPKs) and cholangiocarcinoma: the missing link. Cells. 2019;8(10):1172. doi:10.3390/cells8101172
- Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651-662. doi:10.1016/S1470-2045(16)00078-4
- Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2020;6:160. doi:10.3389/fmolb.2019.00160
- Mino M, Kanno K, Okimoto K, et al. Periostin promotes malignant potential by induction of epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatol Commun. 2017;1(10):1099-1109. doi:10.1002/hep4.1114
- Utispan K, Sonongbua J, Thuwajit P, et al. Periostin activates integrin α5β1 through a PI3K/AKT‑dependent pathway in invasion of cholangiocarcinoma. Int J Oncol. 2012;41(3):1110-1118. doi:10.3892/ijo.2012.1530
- Van Cutsem E, Tempero MA, Sigal D, et al. Randomized phase iii trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol. 2020;38(27):3185-3194. doi:10.1200/JCO.20.00590
- Wu MJ, Shi L, Dubrot J, et al. Mutant IDH inhibits IFNγ-TET2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov. 2022;12(3):812-835. doi:10.1158/2159-8290.CD-21-1077
- Rodrigues PM, Vogel A, Arrese M, Balderramo DC, Valle JW, Banales JM. Next-generation biomarkers for cholangiocarcinoma. Cancers (Basel). 2021;13(13):3222. doi:10.3390/cancers13133222
- Liu J, Ren WX, Shu J. Multimodal molecular imaging evaluation for early diagnosis and prognosis of cholangiocarcinoma. Insights Imaging. 2022;13(1):10. doi:10.1186/s13244-021-01147-7
- Wheatley RC, Kilgour E, Jacobs T, et al. Potential influence of the microbiome environment in patients with biliary tract cancer and implications for therapy. Br J Cancer. 2022;126(5):693-705. doi:10.1038/s41416-021-01583-8










Adapting to a Robotic Workstation for Image-Guided Liver Cancer Surgery
December 4th 2023Govindarajan Narayanan, MD, speaks to the potential time-saving advantages of using the Epione robot for microwave ablation, cryoablation, and other surgical strategies in patients with liver cancer and other tumors.





