Novel and Expanded Oncology Drug Approvals of 2016-PART 1: New Options in Solid Tumor Management
This article focuses on the new agents and indications that emerged in 2016 for solid tumor treatment. We review the drug indications, mechanisms of action, pivotal trial data, pertinent toxicities, use in special populations, and the appropriate clinical contexts for treatment planning.


Oncology (Williston Park). 31(2):110-121.


Table 1. 2016 FDA Approvals of Anticancer Therapies for Solid Malignancies
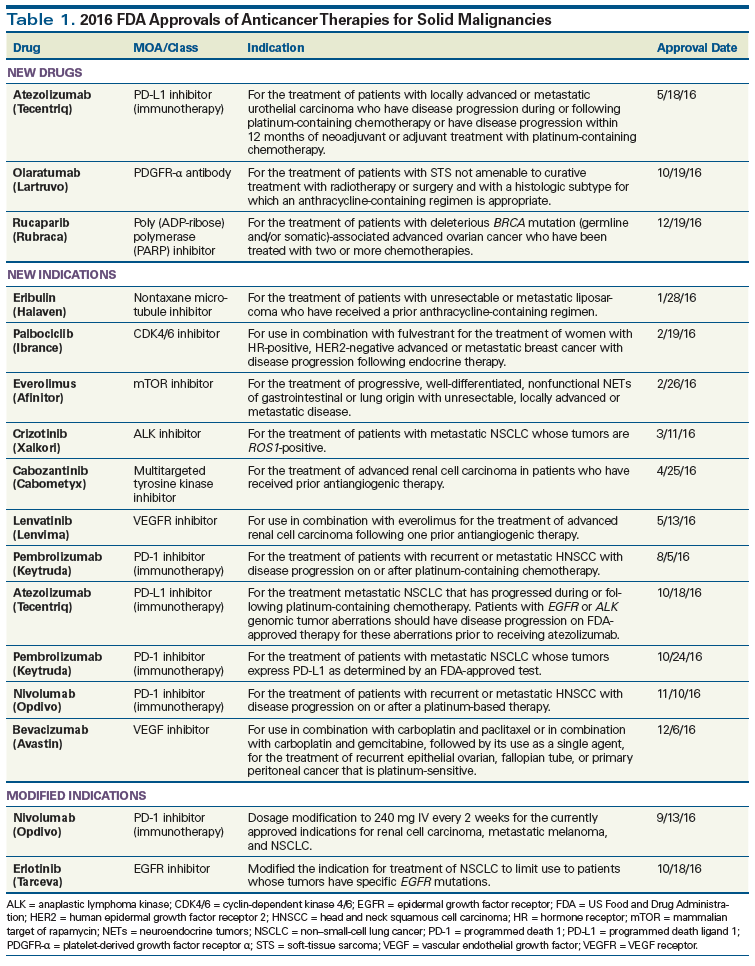


Table 2. FDA Expedited Program/Designation Leading to New Approved Anticancer Therapies for Solid Tumors in 2016
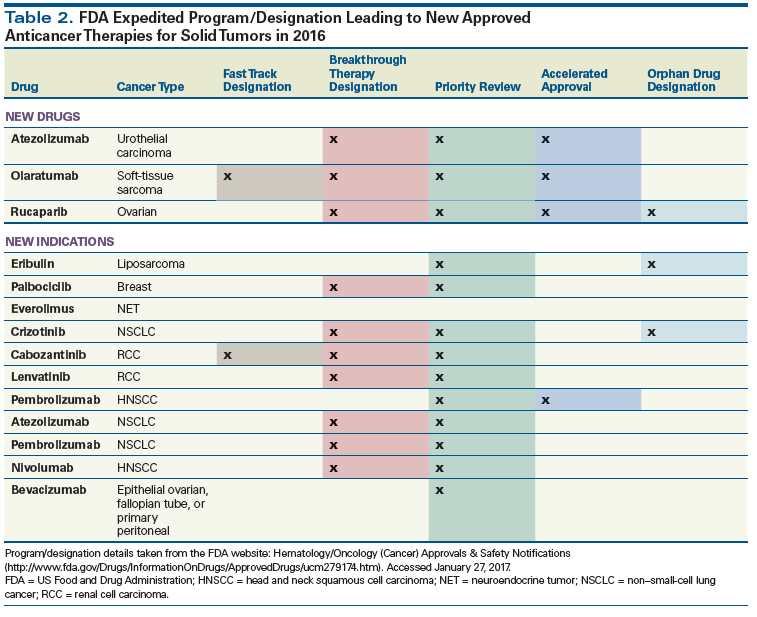
The nonradiologic medical management of solid tumors has evolved from the use of traditional cytotoxic agents to modern targeted therapies, monoclonal antibodies, and immunotherapies. Advances in the understanding of cancer biology and therapeutic strategies have resulted in increasing numbers of new drug applications and approvals. Consequently, practicing oncologists need to learn how the newly available agents function and what toxicities to watch for, as well as ways to optimize the use of both new drugs and previously approved drugs with new indications. In 2016, the US Food and Drug Administration approved three novel drugs for the treatment of solid malignancies-olaratumab in selected patients with soft-tissue sarcoma, atezolizumab for the treatment of bladder cancer, and rucaparib for the treatment of ovarian cancer; also in 2016, the use of previously approved anticancer agents (including atezolizumab) was expanded into 11 new patient populations. The diversity of options for patients is evident in the broad range of the 2016 approvals, which include immune checkpoint inhibitors, targeted therapies, monoclonal antibodies, and traditional cytotoxic agents. This article focuses on the new agents and indications that emerged in 2016 for solid tumor treatment. We review the drug indications, mechanisms of action, pivotal trial data, pertinent toxicities, use in special populations, and the appropriate clinical contexts for treatment planning.
Introduction
Advances in our understanding of the cancer genome and our ability to harness the patient’s immune system to fight cancer continue to translate into US Food and Drug Administration (FDA) approval of new agents for the treatment of cancer. In 2016, three novel drugs were approved to treat patients with solid malignancies; in contrast, 16 new molecular entities were approved in 2015 and 9 were approved in 2014.[1,2] Among the new approvals is the first-in-class programmed death ligand 1 (PD-L1) inhibitor atezolizumab, which was also granted a second indication in 2016. In addition to the two new approvals for this novel agent, two new indications have emerged for the previously approved programmed death 1 (PD-1) inhibitor pembrolizumab, and one new indication has emerged for nivolumab, highlighting that immunotherapy continues to transform cancer care. There were a total of 11 new indications that expanded the use of previously approved anticancer agents into new patient populations (Table 1).
Sponsors of oncology drugs continued to make use of the FDA’s expedited approval programs as pathways to approval in 2016 (Table 2). The Breakthrough Therapy designation is granted to drugs that, based on preliminary clinical evidence, may yield substantial improvement of a clinically significant endpoint, compared with other available therapies. Among the 2016 approvals in solid malignancies, 10 of 14 (71%) were granted Breakthrough Therapy designation, including all three of the new drugs. The Priority Review designation is granted for drugs that, if approved, would provide a significant improvement over the standard-of-care therapies in terms of treatment safety or effectiveness. Priority Review was granted to 13 of 14 approvals (93%), including all three of the novel drugs granted Breakthrough Therapy designation.
In this article we review the three newly approved drugs indicated for use in the oncology population and discuss the new indications of already approved agents. We will focus on pivotal trial data for each agent, as well as the drug’s indication, mechanism of action, pertinent toxicities, use in special populations, and the appropriate clinical context for treatment planning.
New Drug Approvals
Olaratumab, for soft-tissue sarcoma
Olaratumab is a human immunoglobulin (Ig) G1 antibody directed against platelet-derived growth factor receptor α (PDGFR-α). This novel agent received accelerated FDA approval on October 19, 2016 for advanced soft-tissue sarcoma, and had already been granted Priority Review status in May 2016. The drug is specifically approved in combination with doxorubicin for adult patients with soft-tissue sarcoma subtypes for which an anthracycline-based regimen is appropriate and for which curable options are not available. Treatment options are limited for soft-tissue sarcoma and typically involve doxorubicin, administered either alone or in combination. Doxorubicin was combined with olaratumab in the pivotal trials.
The accelerated approval of olaratumab was based on the results from an open-label phase Ib trial and randomized phase II trial of adult patients with locally advanced or metastatic soft- tissue sarcoma who had not previously received doxorubicin. The phase Ib portion enrolled 15 patients who received olaratumab at a dose of 15 mg/kg IV on days 1 and 8 concurrently with doxorubicin at 75 mg/m2 on day 1 of each 21-day cycle, for a maximum of 8 cycles (with dexrazoxane permitted for cardioprotection), followed by continuation of olaratumab maintenance, until disease progression. The phase II portion of the trial randomized 133 patients in a 1:1 ratio to either the combination using the phase Ib dosing regimen or to single-agent doxorubicin. Patients who initially received single-agent doxorubicin could later receive single-agent olaratumab following evidence of disease progression. The primary endpoint of the phase Ib trial was safety, and for the phase II trial was progression-free survival (PFS). The phase II trial was designed with a planned sample size of 130 patients to have 80% power and a two-sided significance level of 0.20 that was adjusted to 0.1999 following the planned interim analysis. The most common soft-tissue sarcoma histology in this group was leiomyosarcoma (36%), followed by undifferentiated pleomorphic sarcoma (15%), liposarcoma (12%), and angiosarcoma (6%). The median PFS in the phase II portion favored the combination of olaratumab and doxorubicin (6.6 months) compared with doxorubicin alone (4.1 months; stratified hazard ratio [HR], 0.67; 95% CI, 0.44–1.02; P = .0615). More notably, the median overall survival (OS) was significantly improved by 11.8 months in the patients who received combination therapy (OS, 26.5 months) compared with patients randomized to doxorubicin alone (OS, 14.7 months; stratified HR, 0.46; 95% CI, 0.30–0.71; P = .003). This survival benefit was consistent across histologic subtypes, number of prior therapies, and PDGFR-α status. There was no correlation between response and tumor positivity for PDGFR-α.[3]
Grade 3/4 adverse events (AEs) associated with doxorubicin and olaratumab included neutropenia (53%), leukopenia (36%), anemia (13%), and fatigue (9%). ANNOUNCE (ClinicalTrials.gov identifier: NCT02451943) is an ongoing phase III randomized trial of doxorubicin and olaratumab in combination vs doxorubicin plus placebo that will help to validate the results of previous trials.
Atezolizumab, for the treatment of bladder cancer and non–small-cell lung cancer (NSCLC): two approvals
Atezolizumab is an anti–PD-L1 monoclonal IgG1 antibody. It was granted both first approval and a new indication by the FDA in 2016, and is the first FDA-approved PD-L1 inhibitor. The initial indication was for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression either during or following platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. In October 2016, atezolizumab joined nivolumab and pembrolizumab as approved immune checkpoint inhibitors for treatment of metastatic NSCLC that had progressed during or following platinum-based chemotherapy. Atezolizumab was also approved in advanced NSCLC that has progressed during or after treatment with epidermal growth factor receptor (EGFR) inhibitors or anaplastic lymphoma kinase (ALK) inhibitors. The FDA also approved the complementary VENTANA PD-L1 (SP142) immunohistochemistry assay for patients with metastatic urothelial cancer or NSCLC who are candidates for treatment with atezolizumab.[4-6] Although PD-L1 expression is not required for initiation of therapy, higher levels of expression are associated with an increased likelihood of response to atezolizumab.
As an immune checkpoint inhibitor, atezolizumab selectively binds PD-L1, preventing interaction with PD-1 and CD80 (also called B7-1), while sparing interaction between programmed death ligand 2 (PD-L2) and PD-1. This ultimately results in inhibition of T-cell suppression while potentially preventing autoimmunity, which would reduce the occurrence of immune-related AEs.[7,8] The mechanism of PD-L1 inhibition by atezolizumab differs from that of previously approved immunotherapies that block PD-1 (nivolumab and pembrolizumab) and cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4; ipilimumab), although there is overlap in efficacy and toxicity.
Atezolizumab in bladder cancer. The study supporting FDA approval of atezolizumab in urothelial carcinoma was a single-arm, multicenter, phase II trial of 310 patients with locally advanced or metastatic bladder cancer who had previously been treated with platinum-based therapy.[4,9,10] Atezolizumab was dosed at 1,200 mg IV on day 1 of each 21-day cycle. The primary endpoint was the objective response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 and the immune-modified RECIST criteria (irRECIST).[11] Patients were prospectively stratified by levels of PD-L1 expression. In comparison with a historic ORR of 10%, the ORR was 15% (P < .0058), and higher response rates correlated with higher levels of PD-L1 expression. The 12-month OS rate was 29% in patients with no PD-L1 expression and 48% in those with the highest expression. After a median follow-up of 11.7 months, 84% of responders had ongoing responses, with complete responses (CRs) in 5.5% of patients overall.[4] In conjunction with an exploratory outcome evaluation, this study also used a targeted exome-sequencing panel to correlate the degree of mutational load as a predictor of atezolizumab response. The median mutational load was 12.4 mutations/Mb in the responders compared with 6.4 mutations/Mb in the nonresponders (P < .0001), results similar to outcomes reported for pembrolizumab-treated patients with melanoma.[12]
Atezolizumab in NSCLC. The pivotal POPLAR and OAK trials were the basis for FDA approval of atezolizumab in NSCLC. The POPLAR trial was a phase II randomized controlled trial of 287 patients with NSCLC who progressed on post-platinum chemotherapy.[13] Patients were randomized in a 1:1 ratio to atezolizumab at a dosage of 1,200 mg IV every 3 weeks or docetaxel at 75 mg/m2 every 3 weeks. They were stratified based on the status of PD-L1 tumor-infiltrating immune cells, histology, and number of previous lines of therapy. The primary endpoint was OS in the intention-to-treat population and PD-L1 subgroups. The median OS durations in the POPLAR trial were 12.6 months and 9.7 months for the atezolizumab and docetaxel arms, respectively (HR, 0.73; 95% CI, 0.53–0.99; P = .04). Patients with nonsquamous disease had increased OS durations-15.5 months and 10.9 months for the atezolizumab and docetaxel arms, respectively-compared with those who had squamous disease, for whom OS was 10.1 months and 8.6 months in the two arms, respectively. PFS was similar, at 2.7 months and 3.0 months for the atezolizumab and docetaxel arms, respectively.
The OAK trial enrolled 1,225 patients with advanced NSCLC who had progressed on a prior platinum-containing regimen, or had experienced disease recurrence within 6 months of treatment with a platinum-based adjuvant/neoadjuvant regimen or combined modality with curative intent. Enrolled patients were randomized 1:1 to atezolizumab (at 1,200 mg IV every 3 weeks) or docetaxel (at 75 mg/m2 every 3 weeks). The median OS duration was 13.8 months in the atezolizumab arm compared with 9.6 months in the docetaxel arm (P = .0003). Improvement in OS was seen regardless of PD-L1 expression; however, OS was significantly greater in the groups with the highest PD-L1 expression (median OS, 20.5 months with atezolizumab vs 8.9 months with docetaxel; HR, 0.41; P < .0001). In 425 intention-to-treat patients, the ORRs in the atezolizumab vs the docetaxel arms were similar at 13.6% and 13.4%, respectively. However, the duration of response was 16.3 months with atezolizumab vs 6.2 months with docetaxel. The PFS durations were 2.8 months in the atezolizumab arm vs 4.0 months in the docetaxel arm.[14]
Within the POPLAR, OAK, and urothelial carcinoma trials, atezolizumab was well tolerated; consistent with previous studies, its safety profile was favorable in comparison with docetaxel.[14] In the pivotal trial for urothelial carcinoma, AEs were reported in 16% of all patients treated with atezolizumab. Fatigue was the most common grade 3/4 AE (2% of patients). Immune-related AEs reported in at least 5% of patients included pneumonitis, elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels, rash, and dyspnea. Among patients treated with atezolizumab, 4% discontinued therapy because of AEs.[4] Within the POPLAR trial, in patients treated with atezolizumab, the most common grade 3/4 AE (≥ 2%) was dyspnea (~ 9%). Significant immune-related AEs for patients receiving atezolizumab included pneumonitis, hepatitis (elevated AST and ALT), and colitis. Among patients treated with atezolizumab, 8% discontinued therapy because of AEs, vs 22% of patients treated with docetaxel. Within the OAK trial, grade 3/4 AEs occurred in 15% of patients treated with atezolizumab, with no associated deaths. The FDA warnings/precautions in atezolizumab product labeling concern immune-mediated reactions that are class-
specific to immunotherapy.
Atezolizumab represents the first approved anti–PD-L1 therapy, a new member of the evolving group of immunotherapy options for a variety of malignancies, including bladder and lung cancer. Individualized clinical information, such as strength of PD-L1 positivity, may guide the decision of whether or not atezolizumab should be considered as treatment; there also may be situations in which caution should be exercised-for example, in patients with autoimmune disease.[15] The search for clinically useful biomarkers to help determine who is most likely to benefit from these immune therapies is ongoing, with increasing consideration of their relative costs and toxicities.
Rucaparib, new approval for pretreated ovarian cancer patients with BRCA mutations
Rucaparib is a novel inhibitor of the enzyme poly (ADP-ribose) polyermase (PARP), which is involved in DNA repair through mechanisms that include base excision repair. The genes BRCA1 and BRCA2 encode tumor suppressors also involved in DNA repair through homologous recombination.[16] The PARP inhibitors exert their effect through synthetic lethality, in which a mutation in just one gene (ie, BRCA1) does not result in cell death, but inhibition of both pathways of DNA repair (ie, base excision repair via BRCA1 and homologous recombination via PARP) and will result in cell death.[17] Patients with BRCA1 or BRCA2 mutations in either their germline or tumor samples have been shown to have increased sensitivity to inhibitors of PARP.
Rucaparib received accelerated FDA approval on December 19, 2016 for patients with advanced ovarian cancer who had received therapy with two or more chemotherapy agents and who also have deleterious germline and/or somatic BRCA mutations. To determine the presence of a deleterious mutation in BRCA1 or BRCA2, a required companion diagnostic test was also approved. The FoundationFocus CDxBRCA test utilizes next-generation sequencing to determine the presence of insertion alterations > 4 nucleotides and deletions > 10 nucleotides in either BRCA1 or BRCA2 in formalin-fixed, paraffin-embedded ovarian tumor tissue, without distinguishing between germline vs somatic mutations.[18]
The evidence supporting this accelerated approval was based on two multicenter, phase II, single-arm trials in 106 patients with advanced BRCA-mutant ovarian cancer following progression after two or more prior platinum-based chemotherapy regimens. The majority of patients had deleterious germline BRCA mutations, although 17% had somatic mutations only. Patients received rucaparib at 600 mg orally each day. The ORR was 54%, with 9% CRs and 45% partial responses (PRs). The median duration of response was 9.2 months.[19] In a population of 206 patients enrolled in the ARIEL2 trial, the most common grade 3/4 toxicities were anemia and elevated levels of liver enzymes.[20]
Rucaparib now joins the approved PARP inhibitor olaparib for the treatment of advanced ovarian cancer. Whereas olaparib was specifically assessed and approved for patients with deleterious germline BRCA mutations, rucaparib is approved for patients with both germline and/or somatic mutations. Additional trials are ongoing to further define the role of PARP inhibitors in cancer management, including assessment of rucaparib in the treatment of ovarian cancer and other cancers related to alterations in DNA damage pathways.
New Indications for Already Approved Drugs
In addition to newly approved drugs, several agents that were previously approved by the FDA received additional indications in 2016. These include the immune checkpoint inhibitors pembrolizumab and nivolumab; the cytotoxic agent eribulin; the monoclonal antibody bevacizumab; and the oral targeted inhibitors crizotinib, cabozantinib, lenvatinib, palbociclib, and everolimus. These expanded approvals are reviewed below.
Immunotherapy
Expanded approvals of anti–PD-1 agents: pembrolizumab and nivolumab in head and neck squamous cell carcinoma (HNSCC) and pembrolizumab in NSCLC. Pembrolizumab and nivolumab are anti–PD-1 monoclonal IgG4 antibodies. Pembrolizumab was previously approved for the treatment of metastatic melanoma, and nivolumab was previously approved for the treatment of patients with metastatic melanoma, NSCLC, and renal cell carcinoma. In 2016, the approved indications for each of these new agents were expanded into new patient populations. Pembrolizumab was approved for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy, and for two new indications in the treatment of patients with metastatic NSCLC.
One new indication for pembrolizumab is for its use as initial therapy in patients whose tumors express PD-L1 with a tumor proportion score (TPS) ≥ 50%, and who are ALK/EGFR-negative and have not received prior systemic chemotherapy. The other indication is for use as subsequent therapy in patients whose tumors express PD-L1 with a TPS ≥ 1%, and who have disease progression during treatment with a platinum-containing chemotherapy regimen (or anti-ALK/EGFR therapy in patients with those genetic mutations). Prior to administration of pembrolizumab, TPS must be determined by an FDA-approved test such as the concurrently approved companion PD-L1 IHC 22C3 pharmDx assay.[21,22] Nivolumab was approved for the treatment of recurrent or metastatic HNSCC with disease progression on or after platinum-based chemotherapy.[23]
Pembrolizumab and nivolumab block PD-1, preventing the interaction between PD-1 and its ligands PD-L1 and PD-L2. Blocking the PD-1/PD-L1 pathway prevents immune suppression of cytotoxic T cells. This therapy may be particularly effective in tumor types that rely on upregulation of the PD-1/PD-L1 pathway as a survival strategy.[21,24,25]
The approval of pembrolizumab in patients with metastatic HNSCC was based on the demonstration of a durable ORR in KEYNOTE-012, a nonrandomized, open-label, multicenter study. The trial enrolled 60 patients with recurrent or metastatic HNSCC who had a PD-L1 TPS ≥ 1% and disease progression during or after treatment with platinum-containing chemotherapy. Pembrolizumab was administered at a dosage of 10 mg/kg every 2 weeks. The primary endpoints were ORR and assessment of safety. The ORR was 18% overall, 25% for human papillomavirus–positive patients, and 14% for human papillomavirus–negative patients. After a median follow-up of 14 months, 16% of patients had a PR, 2% had a CR, and 18% had stable disease. The median PFS duration was 2 months. Median OS was 13 months, with 51% of patients surviving at 12 months.[22] Pembrolizumab showed greater effectiveness, as measured by ORR (P = .010) and PFS (P = .020), in patients whose tumors expressed higher levels of PD-L1.[22,26]
Approval of nivolumab in HNSCC was based on results from the phase III CheckMate-141 clinical trial, which showed a longer median OS-7.5 months-with nivolumab at a dosage of 3 mg/kg every 2 weeks IV, compared with a median OS of 5.1 months using investigator’s choice of standard single-agent therapy (HR, 0.70; 97.73% CI, 0.51–0.96; P = .01). Single-agent choices included cetuximab at an initial dosage of 400 mg/m2 IV once, then at 250 mg/m2 IV weekly; methotrexate at 40 mg/m2 IV weekly; or docetaxel at 30 mg/m2 IV weekly. The 1-year OS rate was 36.0% with nivolumab compared with 16.6% with standard therapy. The median PFS with nivolumab was 2.0 months compared with 2.3 months with standard therapy (HR, 0.89; 95% CI, 0.70–1.13; P = .32), with a response rate of 13.3% in the nivolumab group compared with 5.8% in the group that received standard therapy. Grade 3/4 AEs were significantly reduced with nivolumab (13.1%) compared with standard therapy (35.1%). The most common grade 3/4 AEs in the nivolumab group included fatigue (2.1%) and anemia (1.3%), and the most common grade 3/4 AEs in the group treated with standard therapy were neutropenia (7.2%) and anemia (4.5%).[23]
The expanded approvals for pembrolizumab in the lung cancer population were based on several of the KEYNOTE trials. KEYNOTE-001, a dose-escalation phase Ib trial, established a TPS ≥ 50% as the threshold value for PD-L1 expression associated with clinical benefit.[27]
The phase III KEYNOTE-024 trial was designed to assess the benefit of pembrolizumab compared with one of five platinum-based standard first-line chemotherapy regimens in previously untreated stage IV NSCLC without sensitizing EGFR mutations or ALK translocations. All patients were required to have a PD-L1 TPS ≥ 50%. Patients were randomized in a 1:1 ratio to receive treatment with either pembrolizumab at 200 mg IV every 3 weeks for 35 cycles or 4 to 6 cycles of the investigator’s choice of platinum-based combination chemotherapy. The primary endpoint was PFS. Pembrolizumab was associated with a significantly longer median PFS of 10.3 months, compared with 6.0 months in the chemotherapy group. The estimated rate of OS at 6 months was likewise significantly greater among the patients who received pembrolizumab (OS rate, 80.2% with pembrolizumab vs 72.4% in the chemotherapy group). OS was significantly longer in the pembrolizumab group than in the chemotherapy group (HR for death, 0.60; 95% CI, 0.41–0.89; P = .005).[28]
KEYNOTE-010 was designed to assess the benefit of pembrolizumab compared with docetaxel in previously treated patients with stage IV NSCLC. This three-arm phase II/III trial enrolled 1,033 patients whose tumors had a PD-L1 TPS ≥ 1%. Patients were randomized 1:1 to receive treatment with pembrolizumab at two different doses (2 mg/kg or 10 mg/kg, every 3 weeks) or treatment with docetaxel. The primary endpoints were OS and PFS. The median OS durations were 10.4 months, 12.7 months, and 8.5 months for pembrolizumab at 2 mg/kg, pembrolizumab at 10 mg/kg, and docetaxel at 75 mg/m2, respectively. Compared with patients in the docetaxel treatment group, OS was significantly longer for patients treated with pembrolizumab at 2 mg/kg (P = .0008), and at 10 mg/kg (P < .0001). In 442 patients with elevated PD-L1 expression in tumor cells (TPS ≥ 50%), median OS durations were 14.9 months for patients treated with pembrolizumab at 2 mg/kg, 17.3 months for patients who received the 10-mg/kg dose of pembrolizumab, and 8.2 months for patients treated with docetaxel at 75 mg/m2.[29]
KEYNOTE-012 and KEYNOTE-024 both demonstrated acceptable toxicity profiles for pembrolizumab, with fewer AEs than the chemotherapy regimens with which it was compared. In KEYNOTE-012, AEs of any type were seen in 63% of patients with HNSCC treated with pembrolizumab, with the most common being fatigue (22%), pruritus (12%), rash (7%), diarrhea (4%), and musculoskeletal pain (4%). Grade 3 AEs were seen in 17% of patients, the most common of which included elevated AST and ALT levels, and hyponatremia, each seen in 3% of patients.[22,26] In KEYNOTE-024, there was a lower frequency of treatment-related AEs in the pembrolizumab group (73.4%) vs the chemotherapy group (90.0%). The most common grade 3–5 AE was diarrhea (3.9%). Immune-related AEs of any grade occurred in 29.2% of patients, with hypothyroidism (9.1%) being the most common. AEs led to discontinuation of pembrolizumab in 7.1% of patients, compared with 6.0% of patients who received alternate chemotherapy. One treatment-related death occurred in the pembrolizumab group and three deaths occurred in the group treated with alternate chemotherapy.[28]
National Comprehensive Cancer Network (NCCN) guidelines for HNSCC have classified both pembrolizumab and nivolumab as single-agent therapies for use upon disease progression during or after therapy with platinum-containing regimens. Both have shown clinically sigificant activity, and to date there has not been a study directly comparing pembrolizumab with nivolumab.[30] NCCN guidelines for NSCLC classify pembrolizumab as an agent to be used as subsequent therapy for patients with metastatic nonsquamous or squamous cell NSCLC if PD-L1 expression levels are 1% or higher.[31] Pembrolizumab may be used as first-line therapy for patients whose tumors have PD-L1 expression levels of 50% or higher and who are negative for EGFR mutations, ALK rearrangements, and ROS1 rearrangements. HNSCC and NSCLC both have associations with smoking, a contributing factor for high tumor mutation burden, a metric shown to correlate with the effectiveness of immunotherapy.[4,21,29,32,33] Further assessment of tumor mutation burden to identify patients who will derive the greatest benefit from these agents is an ongoing effort.
Small molecules
Palbociclib, approved for advanced or metastatic hormone receptor–positive breast cancer, in combination with fulvestrant. Palbociclib is an orally available inhibitor of cyclin-dependent kinases 4 and 6 (CDK4/6). It was approved by the FDA for use in combination with fulvestrant for the treatment of women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer with disease progression following endocrine therapy.[34] Palbociclib is a first-in-class-and currently the only-FDA-approved inhibitor of CDK4/6. It was initially approved on February 3, 2015 for use in combination with letrozole as initial endocrine-based therapy in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer.
As a CDK4/6 inhibitor, palbociclib exerts its anticancer effects by blocking progression from the G1 phase to the S phase of the cell cycle; the resulting loss of retinoblastoma protein phosphorylation prevents DNA synthesis.[35] HR-positive breast cancers, in particular those that have developed resistance to prior endocrine therapy, have demonstrated reliance upon CDK4/6 activity.[36-38] Based on mechanistic, preclinical, and clinical evidence supporting the synergistic effect of hormonal therapy plus palbociclib in HR-positive breast cancer, the pivotal PALOMA-3 trial was conducted utilizing the estrogen receptor antagonist fulvestrant.[39,40]
PALOMA-3 was a multicenter, double-blind, randomized, phase III study that compared the combination of fulvestrant plus palbociclib at 125 mg daily for 21 consecutive days followed by 7 days off treatment vs fulvestrant plus placebo in women with HR-positive, HER2-negative metastatic breast cancer that had progressed on previous endocrine therapy. The major results were reported across three articles: the first article described the initial results; the second presented a final analysis with extended follow-up, including subgroup and biomarker analyses; and the most recent one provided a detailed safety analysis.[41-43] In total, 521 patients were randomized (2:1) to receive treatment until disease progression or unacceptable toxicity. Stratification was based upon sensitivity to previous hormonal therapy, menopausal status, and presence or absence of visceral metastasis. The primary endpoint was investigator-assessed PFS according to RECIST.[41,42] After a median follow-up of 8.9 months, median PFS was 9.5 months in the combination group compared with 4.6 months in the group randomized to fulvestrant with placebo (HR, 0.46; 95% CI, 0.36–0.59; P < .0001). At the time of publication, the OS data were not fully mature.[42]
The safety of palbociclib plus fulvestrant was evaluated in the 345 patients in PALOMA-3 who were treated with this combination. Neutropenia was the most common AE in this group, with grade 3 neutropenia reported in 55% of patients and grade 4 neutropenia reported in 10%.[43] In total, 34% of patients had at least one dose reduction during the study and 4% of patients discontinued therapy due to AEs.[42] The most common grade 1–4 AEs, other than neutropenia, occurring in at least 10% of patients were leukopenia, infections, fatigue, nausea, anemia, stomatitis, headache, diarrhea, thrombocytopenia, constipation, vomiting, alopecia, rash, decreased appetite, and pyrexia.
Based on the results of PALOMA-3, palbociclib is now indicated, in combination with fulvestrant, for the treatment of women with HR-positive, HER2-negative advanced or metastatic breast cancer who experienced disease progression following endocrine therapy. Clinicians now have a variety of options for the treatment of HR-positive breast cancer, including several endocrine drug therapies that are recommended for patients with recurrent or metastatic disease. The NCCN assigned a category 1 recommendation to use of the palbociclib/fulvestrant combination as endocrine therapy for postmenopausal or premenopausal women with recurrent or metastatic HR-positive, HER2-negative breast cancer who are undergoing ovarian suppression with a luteinizing hormone–releasing hormone agonist, and whose disease had progressed on prior endocrine therapy. While the impact of this novel combination on OS is still to be determined, there is clear consensus that the addition of palbociclib to conventional endocrine therapy represents a significant improvement. The search for biomarkers or other features to identify patient-selection strategies is a focus of ongoing clinical trials.
Eribulin, approved for advanced liposarcoma following treatment with an anthracycline. Eribulin mesylate is a synthetic analog of halichondrin B that inhibits microtubule dynamics, ultimately leading to G2/M-phase cell-cycle blockade and apoptosis. Eribulin’s unique mechanism of action is distinct from the other microtubule inhibitors-including the vincas, taxanes, and epothilones-and early studies indicate that it may maintain activity in disease that has progressed on these agents.[44] This agent was initially approved in November 2010 for the treatment of metastatic breast cancer that had progressed on at least two prior treatment regimens, including an anthracycline and a taxane. On January 28, 2016, eribulin also gained FDA approval for therapy of unresectable or metastatic liposarcoma following treatment with a prior anthracycline-containing regimen.
The evidence supporting approval of eribulin in liposarcoma was from a randomized, open-label, multicenter, phase III trial of eribulin vs dacarbazine in adult patients with previously treated advanced liposarcoma or leiomyosarcoma. Patients were randomized 1:1 to either eribulin at 1.4 mg/m2 IV on days 1 and 8 (n = 228) or dacarbazine (n = 224), with the dosing based on the site of enrollment. They were stratified by disease type (liposarcoma or leiomyosarcoma), number of prior treatment regimens, and geographic region. The primary endpoint was OS, which was improved in patients who received eribulin compared with those receiving dacarbazine (13.5 months vs 11.5 months, respectively; HR, 0.77; 95% CI, 0.62–0.95; P = .0169).
The survival benefit appeared to be limited to patients with liposarcoma, since those treated with eribulin had a median OS duration of 15.6 months, compared with 8.4 months in the patients who received dacarbazine (HR, 0.51; 95% CI, 0.35–0.75; P = .001), although the study was not powered to assess this comparison. Median PFS in the liposarcoma patients was 2.9 months in those treated with eribulin and 1.7 months in those treated with dacarbazine (HR, 0.52; P = .002). In the whole patient population, the median PFS was similar between the treatment groups (2.6 months in each group). Grade 3/4 AEs were rare, with the exception of neutropenia (35%), leukopenia (10%), and anemia (7%). Discontinuation of eribulin secondary to AEs occurred in 8% of patients, and dose reduction was required in 26%.[45]
Part of the rationale for investigating eribulin in the soft-tissue sarcoma population was related to effects of this drug on vascular remodeling that may enhance the tumor response to subsequent chemotherapy.[46] This may explain the OS benefit without clinically meaningful improvement in median PFS observed in the trial discussed previously, since 69% of patients who received eribulin went on to receive subsequent chemotherapies. The phase III approval trial restricted enrollment to liposarcoma and leiomyosarcoma due to evidence from a prior phase II trial demonstrating that only these two sarcoma histologies met the threshold endpoint of at least 30% of patients being progression-free at 12 weeks.[47] The place for eribulin in the treatment of liposarcoma is still being determined in light of the newer approvals of trabectedin and olaratumab, although it will likely remain a later-line option.
Crizotinib, approved for NSCLC with ROS1 rearrangements. Crizotinib is an oral small-molecule tyrosine kinase inhibitor of ALK, ROS1, and the proto-oncogene MET. It was initially approved in January 2011 for patients with metastatic NSCLC whose tumors are ALK-positive; however, given the similarity between the kinase domains of ALK and ROS1, it was also found to inhibit the latter.[48] On March 11, 2016, crizotinib was approved for the treatment of metastatic NSCLC positive for ROS1 rearrangements.
ROS1 rearrangements are rare in NSCLC, occurring in about 1% of all lung adenocarcinomas, with a higher prevalence in younger patients who are nonsmokers. Rearrangements with ROS1 typically involve the same portion of the ROS1 gene fusing with one of at least 11 partners, ultimately resulting in constitutively active ROS1 fusion protein.[49] The initial clinical evidence supporting the use of crizotinib in NSCLC with ROS1 rearrangements was derived from a case report of a 31-year-old male never-smoker, who was found to be ROS1-positive and had near complete response of his multifocal lung tumor at 8 weeks; the patient continued to receive treatment with crizotinib for more than 6 months, with no evidence of recurrence.[49]
The support for the approval of crizotinib in ROS1-positive NSCLC came primarily from an expansion cohort of the original phase I crizotinib trial: 50 adult patients diagnosed with NSCLC who harbored a ROS1 rearrangement. The patients received crizotinib at a dosage of 250 mg by mouth twice daily. The ORR was 72%, with 3 patients having CRs and 33 having PRs. The median duration of response was 17.6 months, and median PFS was 19.2 months. There was no correlation between response and the type of ROS1 rearrangement seen.
Very few grade 3 toxicities were reported, and included neutropenia (10%) and hypophosphatemia (10%).[50] These data are similar to results from the retrospective cohort of 32 ROS1-positive NSCLC patients enrolled in the EUROS1 trial who received the same regimen of crizotinib and demonstrated an ORR of 80%, including 5 CRs.[51] Based on the high rates of response reported, crizotinib is indicated by the NCCN as the preferred first-line therapy for ROS1-positive metastatic NSCLC.[31]
Everolimus, for progressive, nonfunctional gastrointestinal (GI) and lung neuroendocrine tumors. Everolimus is an oral inhibitor of the mammalian target of rapamycin (mTOR), a kinase composed of two protein complexes-mTOR complex 1 and mTOR complex 2-that is activated by numerous upstream receptor tyrosine kinases. Everolimus is an oral allosteric inhibitor of mTOR complex 1.[52] Everolimus is approved for several indications, including progressive neuroendocrine tumors of pancreatic origin. On February 26, 2016, this approval was expanded to include progressive, nonfunctional GI and lung neuroendocrine tumors.
Neuroendocrine tumors originate from the neuroendocrine cells of the body, with about half arising from the GI tract, 27% from the lungs, and only 6% from the pancreas. They are classified as functional if they are associated with hormonal hypersecretion symptoms, or nonfunctional if they are not.[53] Somatostatin analogs, such as octreotide, are effective for malignancies involving hormonal syndromes; however, effective options for the treatment of nonfunctional neuroendocrine tumors, especially in the GI tract and lungs, are very limited. Based on data from studies of pancreatic neuroendocrine tumors and combination trials with octreotide in functional neuroendocrine tumors, everolimus was assessed specifically in patients with nonfunctional neuroendocrine tumors of the GI tract and lungs.[54]
RADIANT-4 was a randomized, double-blind, placebo-controlled, phase III trial of 302 adult patients with advanced, progressive, well-differentiated, nonfunctional neuroendocrine tumors of the lung or GI tract. Patients were randomized 2:1 to standard supportive care and either everolimus at 10 mg by mouth daily or placebo. The primary endpoint was median PFS, which was significantly improved in the patients randomized to treatment with everolimus (PFS, 11.0 months) rather than placebo (PFS, 3.9 months), resulting in a 52% reduction in the risk of progression or death (HR, 0.48; 95% CI, 0.35–0.67; P < .00001). Secondary endpoints included median OS, which had not yet reached maturity, but the first preplanned interim assessment suggested improved survival outcomes; patient assessment is ongoing. The most common grade 3/4 toxicities were stomatitis, diarrhea, and any type of infection. Discontinuation due to AEs was required in 12% of patients, and dose reductions occurred in 67% of patients receiving everolimus.[55]
While everolimus joins other therapies already approved for pancreatic neuroendocrine tumors, the options for neuroendocrine tumors of the lung and GI tract are limited. RADIANT-4 represents the first randomized trial to show therapeutic benefit in this population of patients, especially for those with lung neuroendocrine tumors.
Approved indications for advanced renal cell carcinoma (RCC): single-agent cabozantinib and the combination of lenvatinib and everolimus. Many targeted therapies have been approved for the treatment of advanced RCC since the initial approval of the multitargeted kinase inhibitors sorafenib and sunitinib in December 2005 and January 2006, respectively. Since then, pazopanib, axitinib, everolimus, and bevacizumab (with the latter approved in combination with interferon) have joined the options for systemic therapy in predominantly clear cell RCC.[56] Two additional multitargeted kinase inhibitors, cabozantinib and lenvatinib, are now part of the renal cancer treatment armamentarium. The mechanism of action of these multitargeted kinase inhibitors is believed to be due in part to inhibition of vascular endothelial growth factor receptor (VEGFR), as well as other hypoxia-inducible genes that ultimately are upregulated in RCC as a result of tumor suppressor gene inactivation.[57]
Cabozantinib is a tyrosine kinase inhibitor directed against numerous targets, including RET, MET, VEGFR1–3, and KIT. It was initially approved in May 2012 at a dose of 140 mg by mouth once daily for progressive, metastatic medullary thyroid cancer commonly associated with RET alterations. Cabozantinib at 60 mg by mouth once daily was approved on April 25, 2016 for the treatment of advanced RCC following prior therapy with an antiangiogenic agent like sunitinib or pazopanib.
The FDA approval for cabozantinib in RCC was based on the results of METEOR, an open-label, randomized, phase III trial of 658 adult patients with advanced or metastatic clear cell RCC who had received prior therapy with at least one VEGFR inhibitor. Patients were randomized 1:1 to either cabozantinib at 60 mg daily or everolimus at 10 mg daily and stratified based on the Memorial Sloan Kettering Cancer Center risk group and number of prior therapies with a VEGFR inhibitor. The primary endpoint was median PFS, which was 7.4 months in the cabozantinib-treated patients and 3.9 months in those who received everolimus (HR, 0.51; 95% CI, 0.41–0.62; P < .0001). The ORR also favored cabozantinib compared with everolimus (17% vs 3%; P < .0001). After more than 18 months of follow-up, the final analysis demonstrated that median OS was 21.4 months with cabozantinib and 16.5 months with everolimus (HR, 0.66; 95% CI, 0.53–0.83; P < .00026).[58]
The most common grade 3/4 toxicities were hypertension (15%), diarrhea (11%), and fatigue (9%). Discontinuation due to AEs was required in 9% of patients, and dose reductions occurred in 60% of patients receiving cabozantinib; the latter were most commonly due to diarrhea (16%), hand-foot syndrome (11%), and fatigue (10%).[58,59]
Lenvatinib is a multitargeted tyrosine kinase inhibitor of RET, VEGFR 1–3, fibroblast growth factor receptors 1–4, KIT, and PDGFR-α. It was initially approved in May 2015 for patients with differentiated thyroid cancer who had progressed on radioactive iodine. On May 13, 2016, it was approved for use in combination with everolimus for patients with advanced RCC following treatment with at least one prior antiangiogenic agent. This approval was based on a randomized, three-arm, open-label, phase II trial of 153 adult patients with advanced or metastatic clear cell RCC that had progressed within 9 months of stopping prior therapy with a previous VEGFR-targeted agent (most commonly sunitinib).[60] Patients were randomized 1:1:1 to lenvatinib at 24 mg by mouth daily, everolimus at 10 mg daily, or the combination of lenvatinib at 18 mg daily and everolimus at 5 mg daily. The primary endpoint, median PFS, was significantly improved in the combination group compared with the group receiving single-agent everolimus (14.6 months vs 5.5 months; HR, 0.40; 95% CI, 0.24–0.68; P = .0005), but not in the patients treated with single-agent lenvatinib (median PFS, 7.4 months; HR, 0.66; 95% CI, 0.30–1.10; P = .12).
The most common all-grade side effects seen with the combination of lenvatinib and everolimus were diarrhea, anorexia, fatigue, nausea and vomiting, cough, lipid abnormalities, stomatitis, hypertension, and peripheral edema. The most common grade 3/4 AEs were constipation (37%), diarrhea (20%), fatigue (14%), hypertension (14%), anemia (8%), and nausea and vomiting (6% and 8%). Discontinuation of lenvatinib and everolimus due to side effects occurred in 24% of patients; 71% of patients required a reduced dose of lenvatinib, whereas only 1 patient out of 51 required an everolimus reduction, likely due to the lower 5-mg daily dose of everolimus used in the combination therapy group.[60]
A wealth of first- and second-line therapies are now available for patients with advanced clear cell RCC, and the NCCN has included both of the novel treatment regimens discussed here in their current guidelines for subsequent therapy. The combination of lenvatinib and everolimus is a category 1 recommendation; cabozantinib is also a category 1, preferred subsequent therapy following first-line therapy.[56] Ongoing clinical trials and biomarker elucidation will help us to further refine our evaluation of the numerous treatment options, including immunotherapy agents like nivolumab, as we aim to optimize treatment sequencing decisions.
Monoclonal antibodies
Bevacizumab, for platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer. Bevacizumab is a vascular endothelial growth factor–specific angiogenesis inhibitor. As an inhibitor of the vital process that provides a blood supply to tumors, bevacizumab is indicated for the treatment of various cancers including colorectal cancer, renal cell carcinoma, nonsquamous NSCLC, glioblastoma, and cervical cancer, mostly in combination with other agents. In December 2016, bevacizumab was approved by the FDA for single-agent use following its use in combination with carboplatin and paclitaxel or carboplatin and gemcitabine, for the treatment of platinum-sensitive recurrent epithelial ovarian, fallopian tube, and primary peritoneal cancers. This expands the indication in these cancers, since bevacizumab in combination with chemotherapy was previously approved in 2014 for the platinum-resistant forms of these same cancers.[61]
This new indication was supported by two randomized, controlled phase III studies, OCEANS and GOG-0213. Platinum-sensitive disease was defined as relapse at 6 months or longer following the last treatment with a platinum-based chemotherapy. In the OCEANS study, 484 patients with recurrent ovarian cancer were randomized 1:1 to receive gemcitabine and carboplatin (GC) plus bevacizumab or GC plus placebo. After 6 to 10 cycles, patients received either bevacizumab or placebo, according to treatment arm, until disease progression or unacceptable toxicity. The primary endpoint of PFS showed a 4-month improvement for the GC-plus-bevacizumab arm (12.4 months vs 8.4 months; HR, 0.46; 95% CI, 0.37–0.58; P < .0001). OS was a secondary endpoint; the study was not powered to detect a difference, and OS was shown to be comparable between the two groups. The OS in the GC-plus-bevacizumab arm was 33.6 months, compared with 32.9 months in the control arm (HR, 0.95; 95% CI, 0.77–1.17; P = .65).[61,62] The GOG-0213 trial randomized 673 patients to receive paclitaxel and carboplatin plus bevacizumab for 6 to 8 cycles, followed by bevacizumab maintenance or paclitaxel-plus-carboplatin alone. The primary outcome for this study was OS, which was extended by 5 months in the bevacizumab arm (42.6 months vs 37.3 months; HR, 0.84; 95% CI, 0.69–1.01). The median PFS improvement seen in OCEANS was consistent with results from GOG-0213, which showed an improvement of 3.4 months (13.8 months vs 10.4 months; HR, 0.61; 95% CI, 0.51–0.72).[61]
AEs were consistent with previous trials of bevacizumab. In both of the two studies, grade 3/4 AEs that occurred more frequently (in ≥ 2% of patients) in the bevacizumab arm were hypertension, fatigue, febrile neutropenia, proteinuria, abdominal pain, hyponatremia, headache, pain in extremity, thrombocytopenia, nausea, dyspnea, and epistaxis.[63] In OCEANS, a higher rate of treatment discontinuation for AEs occurred in the bevacizumab arm (23% vs 5%).[62]
In summary, through two large randomized studies, OCEANS and GOG-0213, the PFS benefit of addition of bevacizumab to chemotherapy has been confirmed to extend to platinum-sensitive ovarian cancers. Although OS in GOG-0213 approached statistical significance, this was not achieved. Likewise, in OCEANS, the addition of bevacizumab did not confer an OS benefit. Thus, it is of increased importance to weigh the associated costs and toxicities associated with bevacizumab against the PFS benefit from use of this agent in platinum-sensitive patients.
Conclusion
The growth of the armamentarium of FDA-approved drugs for the treatment of cancer continued into 2016, with 21 novel or expanded approvals (14 in solid tumors and 7 in hematologic malignancies). While the number of novel agents approved declined compared with recent years, the expanded indications for previously approved agents was notable; among these approvals, the immune checkpoint inhibitors have demonstrated efficacy and safety beyond the treatment of melanoma and lung cancer. With several new immune checkpoint agents and targets still in various phases of investigation and development, the rise of immunotherapy in the field of oncology is expected to continue for years to come. In addition, targeted agents, monoclonal antibodies, and novel treatment modalities are ubiquitous across the oncology drug development pipeline.
In the not-too-distant past, treatment selection for patients with cancer was relatively simple. However, the growing list of approved cancer therapies underscores the complexity of patient care in the modern era. With the pace of oncology drug development showing few signs of slowing, oncologists will be faced with issues such as identifying optimized treatment pathways from among a continually expanding suite of options. Clinicians will increasingly have to consider newer regimens in light of traditional efficacy and safety data; and as advances in cancer survival continue, there will also be an increasing need to weigh the cost of a particular therapy against its impact on quality of life.
Financial Disclosure:Dr. Walko has received honoraria from Bristol-Myers Squibb and Merck & Co, Inc. The other authors have no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
References:
1. National Cancer Institute. A good year: FDA approved nine new cancer drugs in 2014. January 23, 2015. https://www.cancer.gov/news-events/cancer-currents-blog/2015/fda-nine-new-cancer-drugs. Accessed January 25, 2017.
2. US Food and Drug Administration. From our perspective: expedited oncology drug approvals. Page last upated February 26, 2016. http://www.fda.gov/Drugs/NewsEvents/ucm482172.htm. Accessed January 25, 2017.
3. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388:488-97.
4. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909-20.
5. Nods for atezolizumab and nivolumab from FDA. Cancer Discov. 2016;6:811.
6. Ventana Medical Systems, Inc. Ventana PD-L1 (SP142) assay. http://www.accessdata.fda.gov/cdrh_docs/pdf16/P160002c.pdf. Accessed February 1, 2017.
7. Ghiotto M, Gauthier L, Serriari N, et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol. 2010;22:651-60.
8. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3-potential mechanisms of action. Nat Rev Immunol. 2015;15:45-56.
9. Ikeda S, Hansel DE, Kurzrock R. Beyond conventional chemotherapy: emerging molecular targeted and immunotherapy strategies in urothelial carcinoma. Cancer Treat Rev. 2015;41:699-706.
10. Sidaway P. Urological cancer: atezolizumab effective against advanced disease. Nat Rev Clin Oncol. 2016;13:266.
11. Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst. 2010;102:1388-97.
12. Daud AI, Wolchok JD, Robert C, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol, 2016. 34:4102-9.
13. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387:1837-46.
14. Barlesi F, Park K, Ciardiello F, et al. Primary analysis from OAK, a randomized phase III study comparing atezolizumab with docetaxel in 2L/3L NSCLC. Ann Oncol. 2016;27(suppl 6):abstr LBA44_PR.
15. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Bladder cancer. Version 1.2017. https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Accessed January 31, 2017.
16. Rouleau M, Patel A, Hendzel MJ, et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293-301.
17. Iglehart JD, Silver DP. Synthetic lethality-a new direction in cancer-drug development. N Engl J Med. 2009;361:189-91.
18. Rucaparib approved for ovarian cancer. Cancer Discov. 2017 Jan 5. [Epub before print]
19. US Food and Drug Administration. Rucaparib. http://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm533891.htm. Accessed January 27, 2017.
20. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75-87.
21. Sul J, Blumenthal GM, Jiang X, et al. FDA approval summary: pembrolizumab for the treatment of patients with metastatic non-small cell lung cancer whose tumors express programmed death-ligand 1. Oncologist. 2016;21:643-50.
22. Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956-65.
23. Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856-67.
24. Wu P, Wu D, Li L, et al. PD-L1 and survival in solid tumors: a meta-analysis. PLoS One. 2015;10:e0131403.
25. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016;9:5023-39.
26. Merck & Co, Inc. Pembrolizumab [prodcut labeling]. Updated October 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125514s012lbl.pdf. Accessed February 1, 2017.
27. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018-28.
28. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823-33.
29. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540-50.
30. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Head and neck cancers. Version 2.2016. https://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf. Accessed January 31, 2017.
31. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Non-small cell lung cancer. Version 4.2017. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed January 31, 2017.
32. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8.
33. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546-58.
34. US Food and Drug Administration. Palbociclib (IBRANCE capsules). http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm487080.htm. Accessed January 9, 2017.
35. Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476-87.
36. Miller TW, Balko JM, Fox EM, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1:338-51.
37. Dean JL, Thangavel C, McClendon AK, et al. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018-32.
38. Thangavel C, Dean JL, Ertel A, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18:333-45.
39. Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
40. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25-35.
41. Turner NC, Ro J, Andre F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209-19.
42. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425-39.
43. Verma S, Bartlett CH, Schnell P, et al. Palbociclib in combination with fulvestrant in women with hormone receptor-positive/HER2-negative advanced metastatic breast cancer: detailed safety analysis from a multicenter, randomized, placebo-controlled, phase III study (PALOMA-3). Oncologist. 2016;21:1165-75.
44. Scarpace SL. Eribulin mesylate (E7389): review of efficacy and tolerability in breast, pancreatic, head and neck, and non-small cell lung cancer. Clin Ther. 2012;34:1467-73.
45. Schoffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387:1629-37.
46. Dybdal-Hargreaves NF, Risinger AL, Mooberry SL. Eribulin mesylate: mechanism of action of a unique microtubule-targeting agent. Clin Cancer Res. 2015;21:2445-52.
47. Young RJ, Woll PJ. Eribulin in soft-tissue sarcoma. Lancet. 2016;387:1594-6.
48. Huber KV, Salah E, Radic B, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014;508:222-7.
49. Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863-70.
50. Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963-71.
51. Mazieres J, Zalcman G, Crino L, et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33:992-9.
52. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868-80.
53. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72.
54. Pusceddu S, De Braud F, Lo Russo G, et al. How do the results of the RADIANT trials impact on the management of NET patients? A systematic review of published studies. Oncotarget. 2016;7:44841-7.
55. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387:968-77.
56. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Kidney cancer. Version 2.2017. https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf. Accessed January 31, 2017.
57. Iliopoulos O. Molecular biology of renal cell cancer and the identification of therapeutic targets. J Clin Oncol. 2006;24:5593-600.
58. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:917-27.
59. Choueiri TK, Escudier B, Powles T, et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1814-23.
60. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-82.
61. Avastin [prescribing information]. Genentech Inc., San Francisco, CA; December 2016. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125085s317lbl.pdf. Accessed January 27, 2017.
62. Aghajanian C, Goff B, Nycum LR, et al. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2015;139:10-6.
63. F. Hoffmann-La Roche Ltd [media release]. Roche’s Avastin (bevacizumab) plus chemotherapy receives FDA approval for platinum-sensitive recurrent ovarian cancer. Basel; December 7, 2016. http://www.roche.com/media/store/releases/med-cor-2016-12-07.htm. Accessed January 27, 2017.









Late Hepatic Recurrence From Granulosa Cell Tumor: A Case Report
Granulosa cell tumors exhibit late recurrence and rare hepatic metastasis, emphasizing the need for lifelong surveillance in affected patients.

