Pancreatic Neuroendocrine Tumor With Humoral Hypercalcemia and High Tumor PD-L1 Score
Abstract: Pancreatic neuroendocrine tumors (NETs) are a heterogeneous group of neoplasms. They can be functioning tumors with secretion of a variety of peptide hormones, or nonfunctioning tumors with metastases to the liver at the time of diagnosis. Well-differentiated tumors tend to be slow-growing and characterized by low tumor mutational burden (TMB) and lower propensity to express PD-L1. Hypercalcemia due to malignancy can occur in about 20% to 30% of patients with cancer. The secretion of parathyroid hormone–related protein (PTH-rP) is among the causes of malignant hypercalcemia and has seldom been associated with hypercalcemia of NETs. Although the therapeutic landscape for neuroendocrine neoplasms has evolved substantially over the past decade, the role of immunotherapy has not yet been completely explored in this group of patients. We present a rare case of a metastatic pancreatic NET with high TMB, high PD-L1 tumor proportion score, and high PTH-rP–related hypercalcemia.


Copur is a medical oncologist/ hematologist at Morrison Cancer Center, Mary Lanning Healthcare in Hastings, Nebraska, and is a professor at the University of Nebraska Medical Center Adjunct Faculty. He is also an Editorial Advisory Board member at ONCOLOGY®.



Vargas is an assistant professor, transplantation surgery, University of Nebraska.

Introduction
Pancreatic neuroendocrine tumors (NETs) are rare. They have an incidence of less than 1 case per 100,000 individuals per year. Pancreatic NETs are generally indolent and carry a much better prognosis than ductal adenocarcinoma of the pancreas.1-3 Although pancreatic NETs account for approximately 1% to 3% of all pancreatic cancers by prevalence, they constitute 10% of all pancreatic cancers due to their better survival.4 The majority (40%-91%) of pancreatic NETs are nonfunctional, but the remainder can present with clinically evident hormonal symptoms depending on the hormones they secrete. Of the functioning tumors, 70% are insulinomas, 15% are glucagonomas, and 10% are gastrinomas and somatostatinomas. The remaining rare functional pancreatic NETs include vipomas and cholecystokinin-secreting tumors.5-7
Hypercalcemia is a well-known complication of cancer and can be caused by local osteolytic effects of cancer, excess 1, 25(OH)2D secretion, or ectopic parathyroid hormone secretion. Humoral hypercalcemia of malignancy, which is caused by the oversecretion of parathyroid hormone–related peptide (PTH-rP), is responsible for 80% of malignancy associated hypercalcemias.8-11 Although humoral hypercalcemia can be caused by any tumor, its presence in patients with pancreatic NETs is extremely rare.12-14
Immunotherapy in oncology has shown promising results in many cancer types. The immune landscape of NETs has yet to be fully defined. Most NETs appear to be mismatch repair proficient and have a low tumor mutational burden (TMB). Only 3% of patients with NETs have been reported to harbor more than 17 mutations per megabase.15 Immune checkpoint molecules are heterogeneously expressed in the surrounding cells of NETs, and it is unclear whether or not tumor-infiltrating lymphocytes may have antitumor effects within the tumor microenvironment. Recently, an immune checkpoint inhibitor combination therapy showed efficacy in patients with high-grade neuroendocrine
carcinoma.16


Wedel is a staff pathologist at Mary Lanning Healthcare.



Merani is an assistant professor, transplantation surgery, University of Nebraska Medical Center.
Case
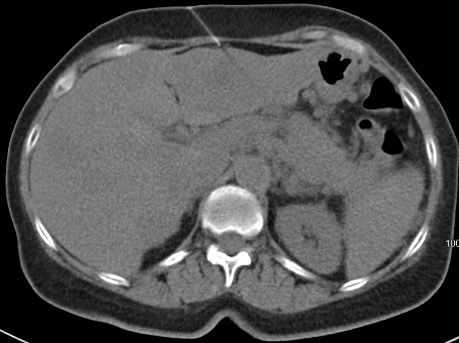
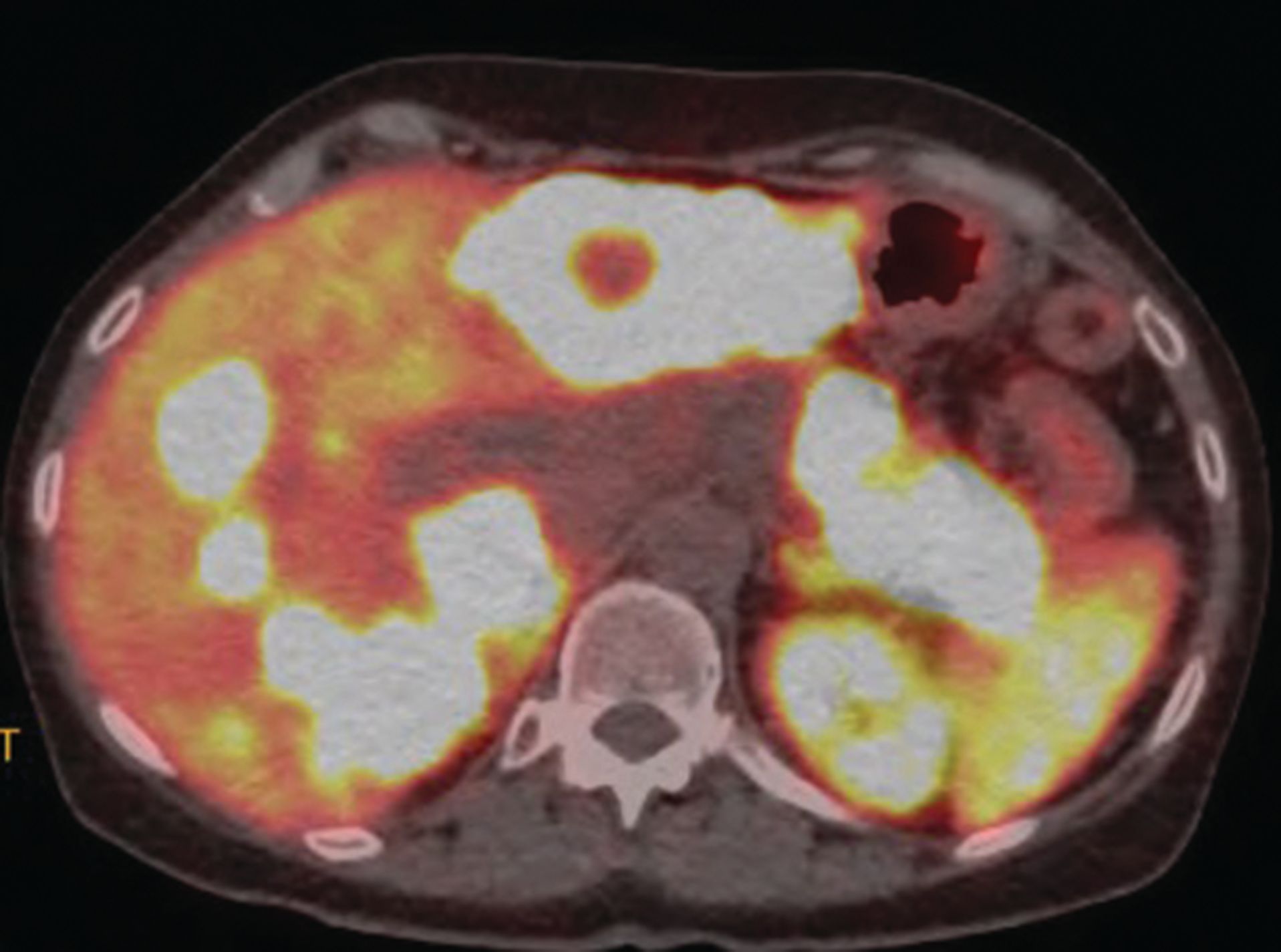
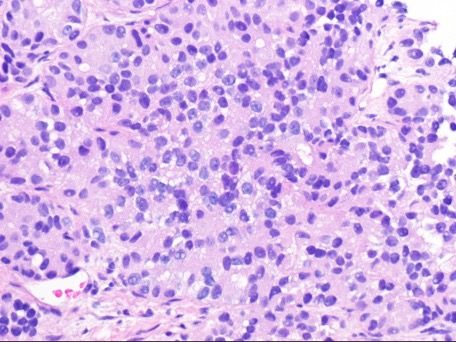
A White woman aged 62 years noted poor appetite, occasional nausea, vomiting, indigestion, and a 30-pound weight loss starting 6 months prior to her diagnosis. Initial evaluation, including upper and lower endoscopies, revealed Barretts esophagus with no dysplasia, a cecal polyp, and negative celiac disease markers. Treatment with proton pump inhibitors did not help her symptoms. As time passed, she noted occasional epigastric and right upper quadrant abdominal pain. Further evaluation with ultrasound and a CT scan of the abdomen revealed a large necrotic mass in the pancreatic body/tail extending to the splenic hilum with invasion into the spleen, as well as multiple enhancing lesions throughout the liver(Figure 1A). A CT-guided biopsy of a liver lesion revealed a metastatic, well-differentiated, high-grade (grade 3) NET with rosette formation, high mitotic rate (more than 20 per high power field), and a Ki-67 index of 30% (Figure 2 A,B). Further work-up with 68gallium 68-dotatate PET-CT confirmed a large pancreatic body/tail PET avid mass with direct extension involving the splenic hilum and multiple hepatic metastases. (Figure 1B). Ultrasound of the thyroid and parathyroid glands andMRI of the brain was negative. Serum gastrin, glucagon, and vasoactive intestinal peptide levels were normal. She had no fasting or nocturnal hypoglycemia. Serum chromogranin A level was elevated at 754 ng/mL (normal 0-103). A genetic consultation and work-up by our certified genetic counselor did not reveal any familial neuroendocrine syndrome.
Shortly after her diagnosis, the patient was noted to have an elevated calcium level of 12.6 mg/dL. Further work-up revealed normal vitamin D 25(OH) (30.69 ng/dL; normal, 30-60); normal vitamin D 1, 25(OH)2 (78 pg/mL; normal, 19-79); suppressed intact PTH (4.6pg/mL; (normal, 12-78); and elevated PTH-rP (32 pmol/L; normal, 0-4.2). Because of the extent of her disease, she was initially treated with systemic fluoropyrimidine-oxaliplatin-based systemic chemotherapy along with monthly long-acting octreotide acetate (Sandostatin LAR Depot) injections. Calcium level returned to normal with intravenous bisphosphonate and isotonic saline. After the completion of 4 cycles of systemic treatment, she underwent an exploratory laparotomy with the intent of tumor debulking, distal pancreatectomy, and splenectomy. However, the extent of tumor invasion of the celiac plexus, left gastric artery, and splenic artery, which were not predictable by the imaging studies, limited the surgery to a wedge resection of the liver.


Cushman-Vokoun is the medical director of the Molecular Diagnostics Laboratory at University of Nebraska Medical Center, and associate professor at the University of Nebraska Medical Center Department of Pathology and Microbiology, with research interest in the molecular pathology and testing of GI-based malignancies.



Drincic is professor of medicine, Division of Diabetes, Endocrinology and Metabolism at the University of Nebraska Medical Center and medical director, Diabetes and Endocrinology Center.

PD-L1 immunohistochemistry analysis of the tumor revealed a tumor proportion score (TPS) of 80%. Next-generation sequencing analysis showed a TMB of 15 per megabase, stable microsatellite status, and genomic findings of RNF43 p.G659fs*41,ATRX p.S750del, BCOR p.L855fs*1,HNF1Ap.P379fs*5, MLH1E172,MSH3 p.K383fs*32,QKI p.K134fs*14. She was started on immune checkpoint inhibitor therapy with pembrolizumab and continued on long-acting octreotide acetate.
Discussion
Our increased understanding of the biology, pathophysiology, and genetics of pancreatic NETs has led to improved diagnostic and treatment options. However, the optimal treatment and sequencing of these treatments remain largely undefined. The relatively uncommon occurrence and indolent nature of some NETs make designing and completing adequately powered randomized trials very difficult. Most patients receive multiple modalities of treatments during the course of their disease. Assessment of the effectiveness of a particular treatment relative to the other treatments becomes very challenging due to the use of a plethora of various treatments with widely varied sequencing. Thus, a lack of established data on optimal treatment sequencing exists. Available practice guidelines offer a wide variety of management options but remain inadequate for the care of individual patients. Involvement of an experienced multidisciplinary team is crucial to provide the optimum coordinated care. For patients with metastatic disease, a multitude of treatment options has ranged from surgical debulking, to systemic chemotherapy, targeted therapy, liver-directed therapy, peptide receptor radionuclide therapy, and more.
Surgery
Surgery is the initial treatment approach for most early-stage tumors. However, patients with incidentally discovered nonfunctioning tumors, especially those less than 1 cm in size, may be followed depending on the site of the tumor.17,18 Resection of the primary tumor and its metastases may be of benefit in selected patients with advanced disease. Metastases in patients with NETs of the pancreas often occur first in the liver. Curative-intent surgical excision of both the primary tumor and the metastases should be considered in patients with limited hepatic disease. This can be performed in a staged or synchronous fashion.19 Noncurative debulking surgery can also be considered in selected cases. When performing staged pancreatoduodenectomy and liver resection, hepatectomy should be considered before pancreatic resection to minimize the risk of perihepatic sepsis from the contaminated biliary tree.20 Additional resection and/or ablation may be possible, providing long-term survival.21 A prophylactic cholecystectomy may be considered, to avoid biliary symptoms and gallstones that could occur due to the continued future use of long-term somatostatin analogues.22


Figure 1A. Large necrotic mass in the pancreatic body/tail extending to the splenic hilum with the invasion into spleen. Multiple enhancing lesions throughout the liver.
Systemic Therapy
Symptomatic patients with clinically significant unresectable tumor burden may benefit from systemic therapy. Several different cytotoxic chemotherapy regimens have been shown to have antitumor activity but no consensus exists on which regimen is the best. Streptozosin is approved for use in patients with advanced pancreatic NETs. The combination of doxorubicin and streptozosinshowed an overall response rate (ORR) of69% and a survival benefit ina small randomized trial.23 An ORR of 39% was reported in a retrospective study using the combination of streptozosin, 5-fluorouracil, and doxorubicin.24 In another study with 88 patients, FOLFOX demonstrated an ORR of 31%, median progression-free survival (PFS) of 9 months, and median overall survival (OS) of 30 months,25 while in a smaller study with FOLFOX, a disease control rate of 78% was observed.26


Figure 1B. Large pancreatic body/tail PET avid mass with direct extension into splenic hilum. Multiple PET avid hepatic metastases.
Octreotide or lanreotide should be considered in certain patients with symptomatic or unresectable disease, high tumor burden, or significant disease progression. Patients with symptoms of hormone secretion or patients who have uptake with somatostatin scintigraphy benefit from somatostatin analogues. Octreotide and lanreotide share the same mechanism of action. In a randomized study of 204 patients with gastroenteropancreatic NETs, treatment with lanreaotide vs placebo resulted in improved PFS (not reached vs 18 months; P <.001).27 Octreotide showed improvement in time to tumor progression (14.3 vs 6 months; P = .000072) in carcinoid tumors of the midgut.28 Radiolabeled somatostatin analogues have shown tumor responses and encouraging survival rates in nonrandomized studies.29,30 In a study involving 610 patients with metastatic gastroenteropancreatic and bronchial NETs, treatment with 177Lu-dotatate resulted in complete responses in 6 patients and a 71-month median OS in patients with pancreatic NETs.31
Oral temozolomide, either alone or in combination, has shown activity in advanced pancreatic NETs. In results of a retrospective series, temozolomide combined with capecitabine showed an objective response rate of 70% and a median PFS of 18 months.32 Kunz et al presented prospective randomized phase 2 trial data comparing single-agent temozolomide with the combination of temozolomide and capecitabine in advanced pancreatic NETs; the combination arm showed improved PFS and OS.33 Ina prospective study combination of temozolomide with bevacizumab, radiographic response was seenin 33% of patients,34 and in another study the combination of temozolomide with everolimus showed partial response in 40% of patients with pancreatic neuroendocrine tumors.35 Everolimus was evaluated in a randomized multicenter study involving 410 patients with advanced progressive pancreatic NETs, and the agent showed PFS benefit compared with placebo (11.0 vs 4.6 months; P <.001).36 Sunitinib was compared with placebo in a randomized study of 171 patients and also showed a PFS advantage over placebo (11.4 vs 5.5 months; P <.001).37
Liver-Directed Therapies
Various liver-directed therapies have been available for hepatic-predominant metastatic disease, mostly to reduce the tumor bulk and relieve symptoms of hormone hypersecretion. Among these are transarterial embolization, chemoembolization, radioembolization, radiofrequency ablation, cryotherapy, and microwave ablation. Embolization in general is considered an effective approach in patients with liver-predominant disease38-40; however, there are limited data comparing various embolization techniques. A systematic review of radiofrequency ablation of 301 patients reported symptom improvement of 92%, with a median duration of 14 to 27 months.41


Figure 2A Pancreatic well-differentiated neuroendocrine tumor. Well-differentiated neoplasm with rosette formation; the Ki67 index was approximately 30% (H&E, 400X magnification).
Microwaves are a nonionizing form of radiation that cause extremely rapid oscillation of the water within tissues; friction from the fluctuation of intracellular water molecules generates heat, which in turn leads to coagulative necrosis. The intratumoral temperatures of microwave ablation are consistently higher than those that can be achieved with radiofrequency ablation. In both radiofrequency ablation and microwave ablation, a probe is placed into the target lesion under radiographic guidance or during open surgery. Multiple lesions can be ablated during the same procedure. However, here is a paucity of data comparing microwave ablation with radiofrequency ablation for management of NETs in the liver.42,43
Cryoablation, another thermoablation technique, works by decreasing cell viability through low temperatures.44 Seifert et al described a series of 13 patients with NETs who underwent hepatic cryotherapy; cryoprobes were inserted under ultrasound guidance. Twelve of 13 patients had complete ablation of all visible tumors, with 2 recurrences at the ablation sites and 12 survivors at 1 year of post-ablation. All 7 patients who had hormonally related symptoms prior to cryotherapy experienced palliative benefit. Two patients developed a postprocedural coagulopathy requiring intraabdominal packing and the transfusion of clotting factors.45


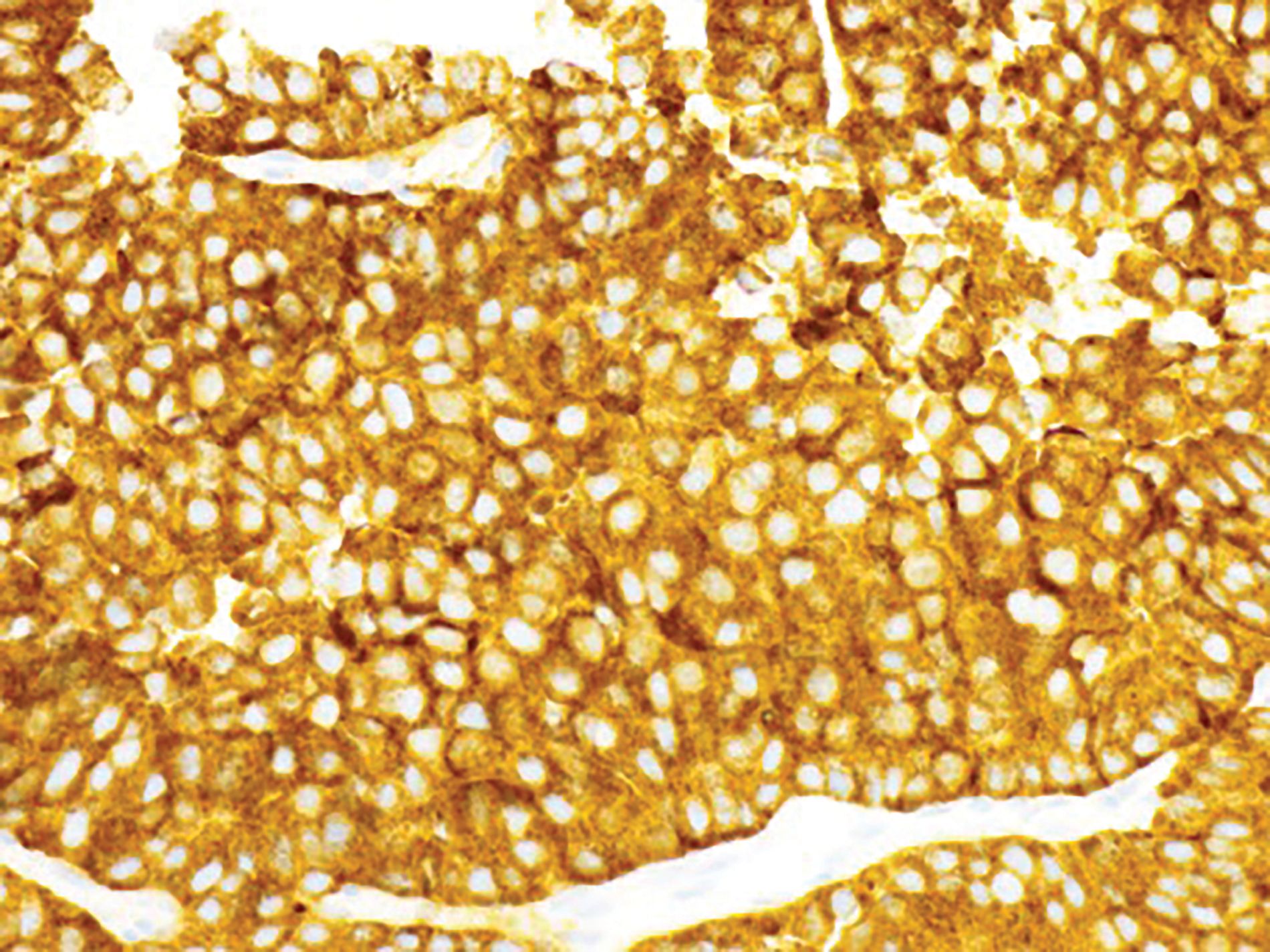
Figure 2B Positivity for synaptophysin (immunoperoxidase stain, 400X magnification).
Unusual Features of the Case
High TMB With a High PD-L1 TPS
PD-L1 immunohistochemistry analysis of the tumor in our patient revealed a TPS of 80%, and next-generation sequencing analysis indicated a TMB of 15 per megabase. The role of immunotherapy in NET patients has not been well defined. Predictive biomarkers such as high TMB and high PD-L1 TPS are being evaluated in identifying patients who might benefit from this treatment modality. Our current knowledge about immunotherapy in gastroenteropancreatic NETs mostly comes from early-phase clinical trials and case reports. In the phase 1b KEYNOTE-028 study, 16 patients with pancreatic NETs who were treated with pembrolizumab monotherapy had an ORR of 6%, PFS of 27% at 12 months, and a medianOS of 87%.46 The analysis of data from a phase 2 basket trial of atezolizumab and bevacizumab in solid tumors, which
included pancreatic and extrapancreatic NET cohorts and involved 40 patients with previously treated grade 1/2 NETs, revealed ORRs of20% and 15% and medianPFS of 19.6 months and 14.9 months in the 2 cohorts, respectively.47 In another phase 2 basket trial, a dual anti–CTLA-4 and anti–PD-1 blockade was tested by utilizing nivolumab plus ipilimumab in 15 patients with nonpancreatic gastrointestinal NETs.16 Objective responses were observed only in high-grade NETs with an ORR of 44%, but the blockade had meager efficacy in the well-differentiated tumors. Several case reports of the use of single-agent or combination immunotherapy in NETshave also been published.48-51 Our patient’s high TMB, high PD-L1 score, and grade 3 histology led us to utilize immunotherapy.
Elevated PTH-rP and Hypercalcemia
An elevated calcium level with a suppressed intact PTH leveland an elevated PTH-rP level was another rather unusual feature of our case. Malignancy-related hypercalcemia is rather uncommon in pancreatic NETs.52-54 PTH-rP is an autocrine, paracrine, and endocrine peptide involving calcium transportation and/or signaling expressed in response to a variety of physiological stimuli.55 It is structurally similar to PTH and, like PTH, it enhances renal tubular reabsorption of calcium while simultaneously increasing urinary phosphorus excretion. However, unlike PTH, it does not increase 1, 25(OH)2 D, thus it does not increase intestinal absorption of calcium and phosphorus. PTH-rP acts on osteoblasts, leading to enhanced synthesis of the receptor activator nuclear kappa B (RANK) ligand.56,57 Bisphosphonates can normalize serum calcium level in more than 70% of patients; however, in approximately 25% of patients, the level can be resistant to bisphosphonate therapy,58 with a relatively short duration (2-3 months) of serum calcium level control. Our patient’s calcium level normalized with intravenous bisphosphonate and isotonic saline; however, her PTH-rP level remained elevated (34 pmol/L) while her intact PTH level remained suppressed (1.4 pg/mL) for 2 months into her hypercalcemia diagnosis and treatment. Serum PTH-rP levels can be considered to be a tumor marker and may indicate treatment response or disease progression.59 PTH-rP has an N-terminal and a C-terminal, which can be utilized as clinically useful assays. While both assays are elevated in patients with humoral hypercalcemia of malignancy, C-terminal PTH-rP depends on glomerular filtration and accumulates in patients with renal failure, while N-terminal PTH-rP remains low or undetectable.60 Our patient had normal renal function. Her elevated but stable PTH-rP level, despite normalization of hypercalcemia, could be indicative of bisphosphonate treatment and stabilization of her disease by the early effect of immunotherapy and octreotide.
Outcome of the Case
Our patient, currently 6 months out from her original diagnosis, is being treated with monthly intramuscular injections of octreotide (Sandostatin LAR 30 mg) and intravenous pembrolizumab 200 mg every 3 weeks. She is currently asymptomatic and tolerating her treatments well. She will be evaluated for further noncurative debulking surgery and/or liver-directed ablative therapy depending on the tumor response to systemic treatment.

References
- Klimstra DS. Nonductal neoplasms of the pancreas. Mod Pathol. 2007;20(Suppl 1):S94-S112.
- Dasari A, Shen C, Halperin D, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335-1342.
- Hallet J, Law CHL, Cukier M, Saskin R, Liu N, Singh S. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer. 2015;121(4):589-597.
- Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19(10):1727-1733.
- Klimstra DS, Arnold R, Capella C, et al. Neuroendocrine neoplasms of the pancreas. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. IARC; 2010:322-326.
- Choti MA, Bobiak S, Strosberg JR, et al. Prevalence of functional tumors in neuroendocrine carcinoma: an analysis from the NCCN NET database. J Clin Oncol. 2012;30(15 suppl; abstr 4126).
- Rehfeld JF, Federspiel B, Bardram L. A neuroendocrine tumor syndrome from cholecystokinin secretion. N Engl J Med. 2013;368(12):1165-1166.
- DeLellis RA, Xia L. Paraneoplastic endocrine syndromes: a review. Endocr Pathol. 2003;14(4):303-317.
- Clines GA, Guise TA. Hypercalcaemia of malignancy and basic research on mechanisms responsible for osteolytic and osteoblastic metastasis to bone. Endocr Relat Cancer. 2005;12(3):549-583.
- Kamp K, Feelders RA, van Adrichem RCS, et al. Parathyroid hormone-related peptide (PTHrP) secretion by gastroenteropancreatic neuroendocrine tumors (GEP-NETs): clinical features, diagnosis, management, and follow-up. J Clin Endocrinol Metab. 2014;99(9):3060-3069.
- Stewart AF, Horst R, Deftos LJ, Cadman EC, Lang R, Broadus AE. Biochemical evaluation of patients with cancer- associated hypercalcemia: evidence for humoral and nonhumoral groups. N Engl J Med. 1980;303(24):1377-1383.
- Kanakis G, Kaltsas G, Granberg D, et al. Unusual complication of a pancreatic neuroendocrine tumor presenting with malignant hypercalcemia. J Clin Endocrinol Metab. 2012;97(4):E627-E631.
- Zhu V, de las Morenàs A, Janicek M, Hartshorn K. Hypercalcemia from metastatic pancreatic neuroendocrine tumor secreting 1,25-dihydroxyvitamin D. J Gastrointestinal Oncol. 2014;5(4):E84-E87.
- Papazachariou IM, Virlos IT, Williamson RC. Parathyroid hormone-related peptide in pancreatic neuroendocrine tumors associated with hypercalcemia. HPB (Oxford). 2001;3(3):221-225.
- Salem ME, Puccini A, Grothey A, et al. Landscape of tumor mutation load, mismatch repair deficiency, and PD-L1 expression in a large patient cohort of gastrointestinal cancers. Mol Cancer Res. 2018;16(5):805-812.
- Patel SP, Othus M, Chae YK, et al. A phase II basket trial of dual anti–CTLA-4 and anti–PD-1 blockade in rare tumors (DART) S1609: the neuroendocrine cohort. Cancer Res. 2019;79(13 suppl; abstr CT039)
- Lee LC, Grant CS, Salomao DR, et al. Small, nonfunctioning, asymptomatic pancreatic neuroendocrine tumors (PNETs): role for nonoperative management. Surgery. 2012;152(6):965-974.
- Strosberg JR, Cheema A, Kvols LK, et al. Stage I nonfunctioning neuroendocrine tumors of the pancreas: surgery or surveillance. J Clin Oncol. 2011;29(4 suppl; abstr 349.
- Lesurtel M, Nagorney DM, Mazzaferro V, Jensen RT, Poston GJ. When should a liver resection be performed in patients with liver metastases from neuroendocrine tumors? a systematic review with practice recommendations. HPB (Oxford). 2015;17(1):17-22.
- De Jong MC, Farnell MB, Sclabas G, et al. Liver-directed therapy for hepatic metastases in patients undergoing pancreaticoduodenectomy: a dual-center analysis. Ann Surg. 2010;252(1):142-148.
- Glazer ES, Tseng JF, Al-Refaie W, et al. Long-term survival after surgical management of neuroendocrine hepatic metastases. HPB (Oxford). 2010;12(6):427-433.
- Oberg K, Kvols L, Caplin M, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15(6):966-973.
- Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D. Streptozosin-doxorubicin, streptozosin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326(8):519-523.
- Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozosin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22(23):4762-4771.
- Girot P, Baudin E, Senellart H, et al. Oxaliplatin and 5-fluorouracil (FOLFOX) in advanced well-differentiated digestive neuroendocrine tumors: a multicenter national retrospective study from the French Group of Endocrine Tumors (GTE). J Clin Oncol. 2019;37(15 suppl; abstr 4104).
- Faure M, Niccoli P, Autret A, Cavaglione G, Mineur L, Raoul J-L. Systemic chemotherapy with FOLFOX in metastatic grade 1/2 neuroendocrine cancer. Mol Clin Oncol. 2017;6(1):44-48.
- Caplin ME, Pavel M, Ćwikla JB, et al; CLARINET Investigators. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
- Rinke A, Müller H-H, Schade-Brittinger C, et al; PROMID Study Group. Placebo-controlled, double blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
- Imhof A, Brunner P, Marincek N, et al. Response, survival, and long-term toxicity after therapy with radiolabeled somatostatin analogue [90Y-DOT]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29(17):2416-2423.
- Kwekkeboom DJ, Bakker WH, Kam BL, et al. Treatment of patients with gastro-entero-pancreatic tumors with the novel radiolabelled somatostatin analogue [177Lu-DOTA (0), Tyr3]octreotate. Eur J Nucl Med Mol Imaging. 2003;30(3);417-422.
- Brabender T, van der Zwan WA, Teunissen JJM, et al. Long-term efficacy, survival, and safety of [177Lu-DOTA0,Tyr3]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Research. 2017;23(16):4617-4624.
- Strosberg JR, Fine RL, Choi J, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117(2):268-275.
- Kunz PL, Catalano PJ, Nimeiri H, et al. A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors: a trial of the ECOG-ACRIN Cancer Research Group (E2211).J Clin Oncol. 2018;36(15 suppl; abstr 4004).
- Chan JA, Stuart K, Earle CC, et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J Clin Oncol. 2012;30(24):2963-2968.
- Chan JA, Blaszkowsky L, Stuart K, et al. A prospective phase 1/2 study of everolimus and temozolomide in patients with advanced pancreatic neuroendocrine tumor. Cancer. 2013;119(17):3212-3218.
- Yao JC, Shah MH, Ito T, et al; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501-513.
- Johnston FM, Mavros MN, Herman JM, Pawlik TM. Local therapies for hepatic metastases. J Natl Compr Canc Netw. 2013;11(2):153-160
- Jones NB, Shah MH, Bloomston M. Liver-directed therapies in patients with advanced neuroendocrine tumors. J Natl Compr Canc Netw. 2012;10(6):765-774.
- Lewis MA, Hubbard J. Multimodal liver-directed management of neuroendocrine hepatic metastases. Int J Hepatol. 2011;2011:452343.
- Mohan H, Nicholson P, Winter DC, et al. Radiofrequency ablationfor neuroendocrine liver metastases: a systematic review. J Vasc Intervent Radiol. 2015;26(7):935-942.e1.
- Ong SL, Gravante G, Metcalfe MS, Strickland AD, Dennison AR, Lloyd DM. Efficacy and safety of microwave ablation for primary and secondary liver malignancies: a systematic review. Eur J Gastroenterol Hepatol. 2009;21(6):599-605.
- Martin RC, Scoggins CR, McMasters KM. Safety and efficacy of microwave ablation of hepatic tumors: a prospective review of a 5-year experience. Ann Surg Oncol. 2010;17(1):171-178.
- Li Y, Wang F, Wang H. Cell death along single microfluidic channel after freeze-thaw treatments. Biomicrofluidics. 2010;4(1):14111.
- Seifert JK, Cozzi PJ, Morris DL. Cryotherapy for neuroendocrine liver metastases. Semin Surg Oncol. 1998;14(2):175-183.
- Ott PA, Bang Y-J, Piha-Paul SA, et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol. 2019;37(4):318-327.
- Halperin DM, Liu S, Dasari A, et al. A phase II trial of atezolizumab and bevacizumab in patients with advanced, progressive neuroendocrine tumors (NETs). J Clin Oncol. 2020;38(4 suppl; abstr 619).
- Stüven AK, Wiedenmann B. Sustained partial remission of a metastatic NEN using off-label immunotherapy with pembrolizumab. Oncotarget. 2019;10(35):3302-3311.
- Ugwu JK, Nwanyanwu C, Shelke AR. Dramatic response of a metastatic primary small-cell carcinoma of the pancreas to a trial of immunotherapy with nivolumab: a case report. Case Rep Oncol. 2017;10(2):720-725. doi:10.1159/000479315
- Schmidt D, Wiedenmann B. Extremely long survival under combined immunotherapy in a metastatic functional neuroendocrine neoplasia patient. Neuroendocrinology. 2018;106(4):381-388.
- Yao JC, Strosberg J, Fazio N, et al. Activity & safety of spartalizumab (PDR001) in patients (pts) with advanced neuroendocrine tumors (NET) of pancreatic (Pan), gastrointestinal (GI), or thoracic (T) origin, & gastroenteropancreatic neuroendocrine carcinoma (GEP NEC) who have progressed on prior treatment (tx). Ann Oncol. 2018;29(suppl 8):viii467-viii468.
- Clemens P, Gregor M, Lamberts R. Pancreatic neuroendocrine tumor with extensive vascularisation and parathyroid hormone-related protein (PTHrP)–associated hypercalcemia of malignancy. Exp Clin Endocrinol Diabetes. 2001;109(7):378-385.
- Rossi RE, Naik K, Navalkissoor S, et al. Case report of multimodality treatment for metastatic parathyroid hormone-related peptide-secreting pancreatic neuroendocrine tumour. Tumori. 2014;100(4):153e-156e.
- Kamp K, Feelders RA, van Adrichem RCS, et al. Parathyroid hormone-related peptide (PTHrP) secretion by gastroenteropancreatic neuroendocrine tumors (GEP-NETs): clinical features, diagnosis, management, and follow-up. J Clin End Met. 2014;99(9):3060-3069.
- Philbrick WM, Wysolmerski JJ, Galbraith S, et al. Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev. 1996;76(1):127-173.
- H Sternlicht, IG Glezerman. Hypercalcemia of malignancy and new treatment options. Ther Clin Risk Manag. 2015;11:1779-1788.
- AE Mirrakhimov. Hypercalcemia of malignancy: an update on pathogenesis and management. N Am J Med Sci. 2015;7(11):483-493.
- Saunders Y, Ross JR, Broadley KE, Edmonds PM, Patel S; Steering Group. Systematic review of bisphosphonates for hypercalcemia of malignancy. Palliat Med. 2004;18(5):418-431.
- Wu TJ, Lin CL, Taylor RL, Kvols LK, Kao PC. Increased parathyroid hormone-related peptide in patients with hypercalcemia associated with islet cell carcinoma. Mayo Clin Proc. 1997;72(12):1111-1115.
- Lum G. Falsely elevated parathyroid hormone-related protein (PTH-RP) in a patient with hypercalcemia and renal failure. Lab Medicine. 2011;42(12):726-728.














