Prostate-Specific Antigen as a Marker of Disease Activity in Prostate Cancer: Part 2


Despite the impact of prostate-specific antigen (PSA) testing on the detection and management of prostate cancer, controversy about its usefulness as a marker of disease activity continues. This review, based on a
ABSTRACT: Despite the impact of prostate-specific antigen (PSA) testing on the detection and management of prostate cancer, controversy about its usefulness as a marker of disease activity continues. This review, based on a recent roundtable discussion, examines whether PSA measurements can be used rationally in several clinical settings. Following radical prostatectomy and radiation therapy, prediction of survival by PSA level is most reliable in high-risk patients. PSA doubling time after radiation therapy is the strongest predictor of biochemical failure. PSA measurements have been associated with inconsistent results following hormonal treatment; reduced PSA levels may result from antiandrogen treatment, which decreases expression of the PSA gene, and therefore, the level of PSA production. In the setting of primary and secondary cancer prevention, PSA is important in risk stratification when selecting patients for studies. Part 2 of this two-part article, which began in the August issue, discusses the role of PSA in hormonal and drug therapies and in primary and secondary chemoprevention. [ONCOLOGY 16:1218-1236, 2002]
In part 1 of this article, which was published in the August issue, we focused on the physiology of prostate-specific antigen (PSA), its measurement and use in clinical practice, and its predictive value following radical prostatectomy and radiation therapy. In part 2, we discuss the role of hormonal and drug therapies in the treatment of prostate cancer and how PSA defines the efficacy of these therapies. We also address the use of various chemopreventive agents such as diet, supplements, and drugs, and consider whether, in evaluating these approaches, an effect on PSA level means a corresponding effect on the disease process.
PSA in Hormonal and Other Drug Therapies
Hormonal therapy is the mainstay of treatment for prostate cancer that has recurred outside the prostate bed and the prostate following surgery or radiation therapy. A variety of medical and surgical options are available that affect points on the hypothalamic-pituitary-gonadal axis. Testicular androgen suppression is achieved with bilateral orchiectomy, estrogen therapy, androgen blockade with a luteinizing hormone-releasing hormone (LHRH) agonist such as goserelin (Zoladex) or leuprolide acetate (Lupron), or an LHRH receptor antagonist such as abarelix depot, which is under investigation in phase III studies.[1]
Additional adrenal androgen suppression (resulting in total androgen ablation) is achieved by adding an oral antiandrogen to testicular ablation; bicalutamide (Casodex), flutamide (Eulexin), and nilutamide (Nilandron) have been approved by the US Food and Drug Administration.
For most patients, these treatments are palliative. Following an initial response, progression (generally to an androgen-insensitive state) becomes manifest.
Variable Results
Reductions in PSA level achieved with hormonal and other therapies in patients with metastatic disease have not always correlated well with survival. For example, studies of combination hormonal therapy have produced inconsistent results. Some clinical trials and meta-analyses of combination therapies have demonstrated a survival benefit over single-agent therapy, with increased survival times ranging from 7 to 20 months,[2] but a meta-analysis of 27 trials revealed approximately a 3% survival benefit at 5-year follow-up for combination hormonal therapy using nonsteroidal antiandrogens.[3]
In some studies, patients treated with hormonal therapy who do not achieve a decline in PSA have worse outcomes than those in whom PSA does decline. Nevertheless, a recent large-scale trial that combined bilateral orchiectomy with flutamide demonstrated no survival benefit with the combination, despite a significantly higher proportion of PSA responses (a reduction in PSA to < 4.0 ng/mL with therapy) in the combination arm (P < .001).[4] In that study, however, the secondary end point-normalization vs no normalization of PSA level following treatment-used 4.0 ng/mL as the cutoff, so the findings did not exclude the possibility that an end point of, say, < 1.0 vs > 1.0 ng/mL might not have been significant.
Because the promoter gene for PSA production is androgen-regulated,[5,6] androgen ablation decreases expression of the gene. It is possible that PSA may not be an appropriate marker in clinical trials in hormone-naive patients,[7] and it is not surprising that certain ranges of PSA decline do not necessarily correlate with clinical outcomes in hormone-treated patients.
FIGURE 1


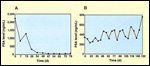
Posttherapy Patterns of PSA Change
Apart from the magnitude of any declines in PSA, the number of times they are documented (minimum should be three) and the specific times during which declines are maintained should be noted. Intermittent declines during periods of chemotherapy only, for example, may be due to transient effects on the synthesis or release of PSA, whereas serial declines toward normal levels and maintenance of low PSA levels may indicate successful treatment (Figure 1).[7] Therefore, it has been strongly recommended that the association between PSA levels and other outcome measures (eg, physical examination, x-rays, scans) continue to be assessed.[8]
In addition, with the exception of generally accepted criteria for measurable disease regression, other parameters used to assess outcome in prostate cancer have not been standardized or validated through association with changes in phase III trials using survival end points. Rather than grouping patient outcomes into a single category such as partial response or stable disease, it would be better if all investigators recorded outcomes based on each disease-related parameter independently.[7] The effect of androgen regulation of PSA is of much less concern when tumors are proliferating despite castrate levels of testosterone.[8]
Monitoring Therapy in Androgen-Independent Cancer
Although PSA levels are quantitatively lower in patients with androgen-independent disease than in patients with androgen-dependent disease,[9] investigators have had greater success in correlating declines in PSA level with survival in androgen-independent disease. Approximately 95% of patients with androgen-independent prostate cancer have elevated PSA levels, and changes in PSA levels often precede changes on bone scans.[7,10]
Several investigators have attempted to correlate a decrease in PSA with clinical benefit and survival, using various agents in androgen-independent prostate cancer. They have noted that a posttherapy PSA decline of 50% is associated with improved survival.[7,8,10] In addition, the interval from elevation of PSA that heralds androgen independence to death is relatively short-the median is about 2 years.[11] In an analysis of 11 different therapeutic protocols at Memorial Sloan-Kettering Cancer Center, a posttherapy PSA decline of 50% or more achieved at 8 and 12 weeks was associated with a significantly improved survival.[7,8]
Based on these studies, a working group of investigators at a consensus conference on PSA assessment of therapy in androgen-independent prostate cancer[10] made the following recommendations:
• In phase II trials, investigators should report a minimum PSA decline of at least 50%, which must be confirmed by a second PSA evaluation performed 4 or more weeks later.
• Additional measures of greater changes in PSA may be reported, and response duration and time to PSA progression may also be important clinical end points.
Participants in this consensus development conference also recommended using PSA to screen for potentially active agents that warrant further study.
Androgen-independent prostate cancer patients constitute a high-risk population that offers the opportunity to validate changes in PSA as a measure of therapeutic efficacy. This can be accomplished by correlating PSA changes with traditional, clinically significant end points such as survival, changes on bone scans, and decreases in cancer-related pain. This can only be done in the context of a prospective randomized trial that demonstrates a survival benefit for a treated (vs a control) population.
Improving Clinical Trial Design
Many phase III trials have failed to confirm the efficacy of therapies that showed great promise in phase I and II studies. To ensure that clinical trials demonstrate the efficacy of a particular therapy for prostate cancer, it will be crucial to detect minimum declines in PSA levels, note the timing of PSA measurements, and validate these findings against clinically significant end points. In addition, phase II and III trials need to be designed differently to improve their ability to demonstrate therapeutic effectiveness.
Conventional phase II trials screen a treatment for any activity against cancer, then estimate antitumor efficacy, and generally accrue about 25 patients using strict entry criteria. Depending on the short-term response to treatment, the investigators decide whether to conduct a more definitive, prospective randomized phase III trial.
Typically, phase III trials of anticancer agents enroll 100 to 300 patients, have more relaxed entry criteria than phase II trials, and use survival as an end point (when the population is at high risk and a survival end point is reasonable). Such a trial may be too small to show a difference in the efficacy of currently available drugs.
Fazzari and colleagues[12] at Memorial Sloan-Kettering Cancer Center have proposed modifying phase II trial designs to make them more comparable to phase III trials. They recommend that phase II trials accrue larger patient cohorts-about 75 patients-possibly involving multiple institutions, with entry criteria that will ultimately be similar to those used for the planned phase III study. In addition, phase II trials should demonstrate sufficient clinical benefit-such as improved survival for high-risk populations-to justify proceeding to a large, randomized trial. The authors view this as the phase II/III transition; ie, what outcome in phase II justifies a phase III study, and of what size?
For example, two ongoing, large-scale trials of cytotoxic agents, docetaxel (Taxotere) vs docetaxel plus estramustine phosphate sodium (Emcyt), and docetaxel plus estramustine vs mitoxantrone plus prednisone, will accrue more than 800 and 600 patients each to demonstrate a 25% and 33% improvement in survival.
• Disease Classification Systems-One issue in trial design is related to disease classification systems. The tumor-node-metastasis (TNM) system poses limitations on the study of prostate cancer, as the disease has a long natural history, most patients are elderly, relapse does not necessarily require a change in therapy, and the likelihood of a non-cancer-related death may exceed that of death from the cancer.
Scher and Heller[13] recently proposed a dynamic staging system for disease ("clinical states") that may address some of these limitations, in that it considers the untreated and treated history of the disease from diagnosis until death. A key aspect of their proposal is the evaluation of a patient’s risk of multiple end points using one database framework. This approach encourages the physician to focus on reducing the risk of death from disease, whether the patient is presenting initially or after a treatment failure. Some patients with a rising PSA after prostatectomy do not die of prostate cancer-an example of "failure" not necessarily equating with death. It is important, therefore, to "reset the clock," reflecting the prognosis at the time of failure and redefining therapeutic objectives.
• Evaluation Standards-No uniform set of standards can be applied to evaluate the effectiveness of drug therapies at all stages of prostate cancer. Although survival is a realistic end point for high-risk patients with metastatic, androgen-independent disease, many therapies may not affect metastatic disease, and may, instead, exert their greatest influence at an earlier stage. Agents that inhibit angiogenesis, for example, may be effective against a growing, localized tumor but not earlier against micrometastatic disease or later against distant metastases.
Moreover, with the advent of increasing numbers of cytostatic drugs, trial design needs to be tailored to the particular characteristics of the responses that may be achieved with these agents; eg, slowing of tumor growth without necessarily inducing detectable tumor regression. This makes the end points (including serum PSA levels) necessary to demonstrate the efficacy of these agents different from those used for cytotoxic drugs. Because the short-term benefits of cytostatic drugs may be difficult to observe or assess, long-term studies of these drugs, with carefully chosen end points, are essential to avoid premature discarding of potentially valuable therapeutic agents.
Transition from one clinical state of disease to another can guide trial design. The goal of treating local disease, for example, is to prevent local or systemic relapse, as evidenced by PSA recurrence or a rising PSA. The goal of treating locally advanced disease is to prevent clinically detectable metastases, as assessed by PSA evidence and negative imaging studies. The goal of treating metastatic but androgen-dependent disease is to prevent transition to androgen independence, as evidenced by rising PSA levels and failure of hormonal therapy.[13,14] Although evaluating drug therapies in prostate cancer will likely depend on clinically significant end points to validate assessment by changes in PSA, those changes in PSA level will play a fundamental role in that assessment.
Several agents that may prevent prostate cancer have been identified, often in epidemiologic studies. Such investigations have pointed to dietary factors, including a low-fat or high-fiber diet, dietary selenium, vitamin E, soy protein, vitamin D, and the carotenoid lycopene, as potential prostate cancer chemopreventives.[15,16] Some of these agents are being studied in prospective trials. The National Cancer Institute and cooperative groups will assess the antioxidants selenium and vitamin E in the Selenium and Vitamin E Cancer Prevention Trial (SELECT), a 12-year randomized prospective study that will enroll more than 32,000 men.[15]
The drug finasteride (Propecia, Proscar), a 5-alpha-reductase inhibitor approved for the treatment of benign prostatic hyperplasia (BPH) and male pattern baldness, is being evaluated in another major study, the Prostate Cancer Prevention Trial (PCPT). This 10-year trial is designed to determine whether finasteride may also prevent prostate cancer.[17,18]
Other promising agents being investigated in prostate cancer chemoprevention include apoptosis inducers (eg, exisulind [Aptosyn]),[19] differentiating agents (eg, vitamin D analogs), antiandrogens (eg, flutamide), antiestrogens (eg, tamoxifen), angiogenesis inhibitors (eg, linomide), antiproliferatives (eg, 2-difluoromethylornithine), and retinoids (eg, vitamin A).[20]
Prostate Intervention Nutrition Study
The end point of prostate cancer chemopreventive studies thus far has been the development of prostate cancer itself. Using PSA as an end point of chemopreventive trials will be meaningful in studies of postprostatectomy recurrence, where it is a clear marker of disease recurrence and activity. One such trial, the Prostate Intervention Nutrition Study, is under way at Memorial Sloan-Kettering Cancer Center.[21]
Patients enrolled in this trial who have developed biochemical recurrence after radical prostatectomy (postsurgical PSA > 0.4 ng/mL and rising on at least two consecutive measurements not exceeding 10 ng/mL) will be randomized to either a diet currently recommended by US guidelines (30% fat) or to one of three dietary interventions. The first intervention will be a diet of 15% fat. The second will be the addition of daily food supplements; ie, 200 µg of selenium (as selenomethionine), 800 IU of vitamin E, and 40 g of soy protein isolate. The third will be a combination of the low-fat diet and supplements.
The end points will be a decrease or stabilization of serum PSA or a decrease in PSA velocity after 1 year. A recently completed study of postprostatectomy recurrence successfully used a similar design to investigate the effect of exisulind on disease progression.[19]
Other Trials of Chemopreventives
In another type of trial designed to evaluate chemopreventives in men with known prostate cancer, investigators will administer potential chemopreventives prior to surgery, and postoperatively assess the molecular, genetic, and histologic characteristics of the tumors, compared with controls. Chemopreventive studies will also be valuable in men who choose watchful waiting instead of definitive therapy. These are often older men with slight evidence of disease on biopsy, small tumor volumes, and low Gleason scores. Annual biopsies and close PSA monitoring will be important in determining the effectiveness of chemopreventives in this group.
Whether PSA can be the major end point of trials of potential chemopreventive agents administered long before prostate cancer develops is less clear. An important consideration in such studies will be whether the agent may affect PSA levels independent of its effect on the disease itself.
Androgens are known to regulate PSA expression in prostate cancer cells. This effect can be blocked by competitive inhibitors of the androgen receptor, such as hydroxyflutamide. For this reason, it may not be appropriate to use PSA as a marker of prostate cancer disease activity in studies of potential chemopreventives that interfere with the androgen receptor.[22]
Prostate Cancer Prevention Trial
The question of an agent affecting PSA without affecting the disease process was a major consideration in the design of the PCPT.[17,18] The effect of finasteride on PSA is dramatic and swift, reducing serum PSA levels by about 50% in 1 year.[17,23]
Finasteride competitively inhibits 5-alpha-reductase, which catalyzes the conversion of testosterone to dihydrotestosterone (DHT). As an androgen, DHT is 10 times more potent than testosterone, with three times more binding power for the androgen receptor. Moreover, DHT is thought to be the principal androgen responsible for both normal and hyperplastic growth of the prostate gland.
The PCPT will test the theory that long-term androgenic stimulation also contributes to prostate carcinogenesis. It is already known that prostate cancer rates are higher among men with high circulating levels of DHT. In addition, neither BPH nor prostate cancer develops in men with an inherited deficiency of 5-alpha-reductase, which results in a mild pseudohermaphroditism but normal serum testosterone levels and virilization at puberty.[15,18] Finasteride seems to inhibit the growth of established prostate cancer cell lines in vitro, and another 5-alpha-reductase inhibitor, turosteride, inhibits tumor growth in prostate cancer xenografts.[15] In addition, in vivo studies in rats have shown that 5-alpha-reductase inhibition can prevent or slow the growth of prostate cancer.[15]
The final determination of whether a 5-alpha-reductase inhibitor may influence PSA levels without affecting prostate cancer itself will have to wait until the completion of the PCPT in 2004. In the meantime, indications are that finasteride does not mask the detection of prostate cancer by PSA when observed PSA values are doubled, as noted in the product labeling.[24]
Until the results of these ongoing studies are known, the end point of chemoprevention trials will likely remain a definitive prostate cancer diagnosis. However, PSA is likely to play a major role in risk stratification when selecting patients for chemoprevention trials.
PSA in Risk Stratification
The long natural history of prostate cancer can be an advantage in studying chemopreventive agents because an agent can be administered before frank prostate cancer develops. To best evaluate a chemopreventive, the subjects should show an increased risk of disease. That fact has led investigators to search for factors other than PSA that would identify patients at risk.
• Prostatic Intraepithelial Neoplasia-High-grade prostatic intraepithelial neoplasia (PIN) is one possible factor. PIN is characterized by proliferation and anaplasia of cells lining the prostatic ducts and acini, but with an intact basal membrane.[25] This histologic feature has long been assumed to be a preinvasive malignant change or a marker of carcinoma. A large autopsy study, however, showed no close geographic or statistical association between high-grade PIN and carcinoma.
Prostate glands of 249 men aged 20 to 69 years who were autopsied at the Wayne County (Michigan) Medical Examiner’s office were studied histologically. Evidence of adenocarcinoma was found in 70 patients, and of high-grade PIN, in 28. In most prostates with carcinoma (49 glands or 70%), high-grade PIN was not present, and in the majority of the 22 prostates that contained both high-grade PIN and carcinoma (15 glands or 68%), these features were geographically separate.[26] Thus, high-grade PIN is unlikely to be a useful risk-stratification tool for chemoprevention.
• Genetic Risk-Another approach to defining high-risk patients for chemoprevention is to identify those with a strong genetic risk. About 9% of prostate cancers result from the inheritance of mutated genes for prostate cancer susceptibility,[27] and about 15% of men with prostate cancer will be found to have a first-degree male relative with prostate cancer.[28] Men with an affected father or brother are twice as likely to develop prostate cancer as men with no affected relatives, and the risk rises with the number of affected relatives. Men with two affected first-degree relatives have a fivefold increased risk, and men with three first-degree affected relatives have an 11-fold increased risk of developing prostate cancer.[28]
Although hereditary cancer is thought to account for 43% of early-onset (age £ 55 years) disease, hereditary prostate cancer is no more virulent than sporadic cancer. Both types show similar clinical stages, preoperative PSA levels, postoperative specimen stages, and prostate weights.[29] For these reasons, men with a family history of prostate cancer would be good candidates for chemoprevention. However, family history will not be the only criterion for selection, because familial clustering may be related to environmental or dietary factors and not necessarily to genetic susceptibility.
• Other Markers-Because moderately elevated PSA levels may indicate BPH or prostatitis, investigators have sought other markers that may be early indicators of prostate cancer carcinogenesis. For example, prostate stem cell antigen (PSCA) is homologous to stem cell antigen 2, which is a marker for the development of hematopoiesis. PSCA is highly expressed in prostatic carcinomas and may prove to be a useful marker.[30]
Although controversial, another proposed marker is insulin growth factor (IGF)-I. High IGF-I levels may correlate with higher rates of epithelial cell proliferation or low rates of apoptosis, and higher IGF-I levels have been associated with prostate cancer in epidemiologic studies. However, levels of IGF-I vary considerably among individuals.[31] Other markers of proliferation, differentiation, angiogenesis, apoptosis, genetic damage, and signal transduction are also currently being investigated.[20]
Recent evidence suggests that BPH, prostatitis, and infection, all of which raise PSA levels, may also be associated with prostate cancer. A study by the National Cancer Institute correlated gonorrhea or syphilis infection with increased risk of prostate cancer and the number of infections with the level of risk (odds ratio = 3.3 among men with three or more events). In chronic inflammatory lesions in the prostate, the epithelium has been shown to be hyperactive and undergoing a dramatic increase in the proliferative rate. De Marzo and colleagues have proposed that this proliferative response renders cells at high risk of DNA damage and the development of neoplasia.[30]
• Lower PSA Levels-PSA levels lower than the usual 4.0-ng/mL cutoff for prostate cancer detection are not incompatible with an increased risk of prostate cancer. The study of PSA levels in patients enrolled in the Physicians Health Study, the Prostate, Lung, Colorectal, and Ovarian cancer screening trial, and the Prostate Cancer Awareness Week trials define men with PSA levels of 2 to 3 ng/mL as being at high risk of progression to high-risk PSA levels within 3 years and to prostate cancer, thereafter. In addition, these studies indicate that men with PSA levels of 1 to 2 ng/mL are also at increased risk of prostate cancer (ie, higher than the risk of men with levels below 1 ng/mL), although their PSA levels do progress more slowly than those of men with levels of 2 to 3 ng/mL.[32,33]
Men in the Physicians Health Study with PSA levels less than 1 ng/mL, however, showed no increased risk of developing prostate cancer. Low PSA levels above 1 ng/mL, therefore, may help define groups of younger men at higher risk of prostate cancer who later may require definitive therapy as candidates for chemoprevention.[34]
• Foci of Prostate Cancer-Some younger men with slightly elevated PSA levels may, in fact, have foci of prostate cancer. An autopsy study of the prostates of young male patients (both African-American and white) aged 10 to 49 years detected small foci of prostate cancer in 27% of men in their 30s and 34% of men in their 40s.[35] This implies that young men with PSA values between 1 and 2 ng/mL may, in fact, have foci of prostate cancer. It also implies that, although it may be difficult to define a group of young men at high risk before the inception of prostate cancer, there is a window of opportunity for chemoprevention between inception and promotion or progression. Biopsies on a fixed schedule will be important in monitoring the effects of chemopreventives in these men.
Identifying groups of younger men at higher risk of prostate cancer will likely involve a combination of risk factors, similar to the Gail criteria,[36] which define cohorts of young women at high risk of breast cancer. Among these factors, PSA will certainly play a dominant role.
References:
1. Doehn C, Jocham D: Technology evaluation: Abarelix, Praecispharmaceuticals. Curr Opin Mol Ther 2:579-585, 2000.
2. Moul JW: Prostate-specific antigen only progression of prostate cancer. JUrol 163:1632-1642, 2000.
3. Prostate Cancer Clinical Trialists’ Collaborative Group: Maximumandrogen blockade in advanced prostate cancer: An overview of the randomisedtrials. Lancet 355:1491-1498, 2000.
4. Eisenberger MA, Blumenstein BA, Crawford ED, et al: Bilaterial orchiectomywith or without flutamide for metastatic prostate cancer. N Engl J Med339:1036-1042, 1998.
5. Riegman PH, Vliestra RJ, van der Korput JA, et al: The promoter of theprostate-specific antigen gene contains a functional androgen responsiveelement. Mol Endocrinol 5:1921-1930, 1991.
6. Wolf DA, Schulz P, Fittler F: Transcription regulation of prostatekallikrein-like genes by androgen. Mol Endocrinol 6:753-762, 1992.
7. Scher HI, Mazumdar M, Kelly WK: Clinical trials in relapsed prostatecancer: Defining the target. J Natl Cancer Inst 88:1623-1634, 1996.
8. Scher HI, Kelly WMK, Zhang Z-F, et al: Post-therapy serumprostate-specific antigen level and survival in patients withandrogen-independent prostate cancer. J Natl Cancer Inst 91:244-251, 1999.
9. Leo ME, Bilhartz DL, Bergstralh EJ, et al: Prostate-specific antigen inhormonally treated stage D2 prostate cancer: Is it always an accurate indicatorof disease status? J Urol 145:802-806, 1991.
10. Bubley GJ, Carducci M, Dahut W, et al: Eligibility and responseguidelines for phase II clinical trials in androgen-independent prostate cancer:Recommendations from the prostate-specific antigen working group. J Clin Oncol17:3461-3467, 1999.
11. Zietman AL, Dallow KC, McManus PA, et al: Time to secondprostate-specific antigen failure is a surrogate endpoint for prostate cancerdeath in a prospective trial of therapy for localized disease. Urology47:236-239, 1996.
12. Fazzari M, Heller G, Scher HI: The Phase II/III transition: Toward proofof efficacy in cancer clinical trials. Control Clin Trials 21:360-368, 2000.
13. Scher HI, Heller G: Clinical states in prostate cancer: Toward a dynamicmodel of disease progression. Urology 55:323-327, 2000.
14. Morris MJ, Scher HI: Novel strategies and therapeutics for the treatmentof prostate carcinoma. Cancer 89:1439-1348, 2000.
15. Brawley OW, Parnes H: Prostate cancer prevention trials in the USA. Eur JCancer 36:1312-1315, 2000.
16. Yip I, Heber D, Aronson W: Nutrition and prostate cancer. Urol Clin NorthAm 26:403-411, 1999.
17. Feigl P, Blumenstein B, Thompson I, et al: Design of the Prostate CancerPrevention Trial (PCPT). Control Clin Trials 16:150-163, 1995.
18. Coltman CA Jr, Thompson IM, Feigl P: Prostate Cancer Prevention Trial (PCPT)update. Eur Urol 35:544-547, 1999.
19. Goluboff E, Prager D, Rukstali D, et al: Safety and efficacy of exisulindfor treatment of recurrent prostate cancer after radical prostatectomy. J Urol166:882-886, 2001.
20. Kelloff GJ, Lieberman R, Steele VE, et al: Chemoprevention of prostatecancer: Concepts and strategies. Eur Urol 35:342-350, 1999.
21. Lee CT, Fair WR: The role of dietary manipulation in biochemicalrecurrence of prostate cancer after radical prostatectomy. Semin Urol Oncol17:154-163, 1999.
22. Crawford ED, DeAntoni E, Ross CA: The role of prostate-specific antigenin the chemoprevention of prostate cancer. J Cell Biochem 255(suppl):149-155,1996.
23. Guess HA, Gormley GJ, Stoner E, et al: The effect of finasteride onprostate-specific antigen: Review of available data. J Urol 155:3-9, 1996.
24. Oesterling JE, Roy J, Agha A, et al: Biologic variability ofprostate-specific antigen and its usefulness as a marker for prostate cancer:Effects of finasteride. The Finasteride PSA Study Group. Urology 50:13-18, 1997.
25. Bostwick DG: Prostatic intraepithelial neoplasia (PIN): Current concepts.J Cell Biochem 16H(suppl):10-19, 1992.
26. Sakr WA, Grignon DJ, Crissman JD, et al: High grade prostaticintraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the agesof 20-69: An autopsy study of 249 cases. In Vivo 8:439-443, 1994.
27. Gronberg H, Isaacs SD, Smith JR, et al: Characteristics of prostatecancer in families potentially linked to the hereditary prostate cancer 1 (HPC1)locus. JAMA 278:1251-1255, 1997.
28. Steinberg GD, Carter BS, Beaty TH, et al: Family history and the risk ofprostate cancer. Prostate 17:337-347, 1990.
29. Carter BS, Bova GS, Beaty TH, et al: Hereditary prostate cancer:Epidemiologic and clinical features. J Urol 150:797-802, 1993.
30. De Marzo AM, Coffey DS, Nelson WG: New concepts in tissue specificity forprostate cancer and benign prostatic hyperplasia. Urology 53(3 suppl 3a):29-39,1999.
31. Pollak M, Beamer W, Zhang JC: Insulin-like growth factors and prostatecancer. Cancer Metast Rev 17:383-390, 1999.
32. Gann PH, Hennekens CH, Stampfer MJ: A prospective evaluation of plasmaprostate-specific antigen for detection of prostate cancer. JAMA 273:289-294,1995.
33. Crawford ED, Chia D, Andriole GL Jr, et al: PSA changes as related to theinitial PSA: Data from the prostate, lung, colorectal and ovarian cancer (PLCO)screening trial (abstract). Proc Am Soc Clin Oncol 20:177a, 2001.
34. Moul JW: Prostate-specific antigen-enhanced testing and riskstratification for chemoprevention trials. Urology 57(4 suppl 1):174-177, 2001.
35. Sakr WA, Haas GP, Cassin BF, et al: The frequency of carcinoma andintraepithelial neoplasia of the prostate in young male patients. J Urol150:379-385, 1993.
36. Gail MH, Brinton LA, Byar DP, et al: Projecting individualizedprobabilities of developing breast cancer for white females who are beingexamined annually. J Natl Cancer Inst 81:1879-1886, 1989.












