Trastuzumab: Mechanisms of Resistance and Therapeutic Opportunities
The human epidermal growth factor receptor 2 (HER2) is a transmembrane receptor with tyrosine kinase activity overexpressed in about 20% to 25% of invasive carcinomas of the breast.
ABSTRACT: ABSTRACT: Many breast cancer patients with HER2 overexpression do notrespond to initial therapy with trastuzumab (Herceptin), and the vastmajority of those who initially respond to the agent develop resistanceto treatment within 1 year. This review will discuss several molecularmechanisms that can lead to the development of trastuzumab resistance,including loss of PTEN, activation of alternative pathways, receptorantibodyinteraction block, and circulating HER2 extracellular domain,as well as the possibility of exploring these aberrations as therapeutictargets that could help avoid or overcome resistance to trastuzumab.
The human epidermal growth factor receptor 2 (HER2) is a transmembrane receptor with tyrosine kinase activity overexpressed in about 20% to 25% of invasive carcinomas of the breast.[1] Patients with such tumors have different responses to therapeutic agents and a worse outlook, with reduced progression-free and overall survival, compared with patients harboring HER2-negative tumors. Trastuzumab (Herceptin) is a humanized IgG1 monoclonal antibody against the extracellular domain of HER2. The combined use of trastuzumab and chemotherapy in randomized trials has resulted in an increase in overall survival rates in the metastatic setting.[2] Furthermore, adjuvant trastuzumab has been shown to reduce the risk of recurrence or death in early-stage breast cancer.[3]
Even though such results have been achieved in patients selected for the presence of HER2 overexpression or gene amplification, a sizable fraction of patients do not respond to initial therapy with trastuzumab, either as a single agent or in combination with chemotherapy, and the vast majority of patients initially responding develop resistance to treatment within 1 year. Such findings suggest that both de novo and acquired mechanisms of therapeutic resistance are important causes of treatment failure.
Because of the complex and redundant signaling pathways of the ErbB family of receptors, which have more than 30 different extracellular domains and more than 50 intracellular effectors, the mechanisms potentially involved in trastuzumab resistance are multiple. In addition, the activation of other receptor families and the abnormal function of their respective signaling pathways, as well as the abnormal activity of several other intracellular molecules that behave as messengers or moderators of these signaling cascades, might also play a role in determining resistance to trastuzumab.
This review will discuss several molecular mechanisms that can lead to the development of trastuzumab resistance, as well as the possibility of exploring these aberrations as therapeutic targets that could help avoid or overcome resistance to trastuzumab, thus enhancing the therapeutic arsenal and the life expectancy of patients with HER2-positive breast cancer.
Postulated Mechanisms of Action of Trastuzumab
Despite the absence of a known direct ligand, the catalytic activity of HER2 is a result of its homo- and heterodimerization with other receptors of the ErbB family, such as the epidermal growth factor receptor (EGFR), HER3, and HER4.[4] Through its binding with high affinity to HER2, trastuzumab is able to block intracellular signaling via the mitogen-activated protein kinase (MAPK) and the phosphatidylinositol 3-kinase (PI3K) pathways.[5,6] The MAPK pathway is critical for tumor cell proliferation, and the PI3K pathway leads to phosphorylation and activation of AKT, with inhibition of apoptosis.
Blocking these pathways leads to the accumulation of the p27Kip1, an inhibitor of cyclin-dependent kinases, thus inhibiting the cyclin E/Cdk2 complex and leading to cell-cycle arrest in G1/S and induction of apoptosis.[7] Trastuzumab therapy also increases membrane localization and activity of PTEN, the protein product of the phosphatase and tensin homolog deleted on chromosome 10 gene, by reducing PTEN tyrosine phosphorylation via Src tyrosine kinase inhibition.[8] Increased PTEN activity thus inhibits the PI3K pathway and cell proliferation.
Immune response activation through antibody-dependent cellular cytotoxicity is another potential mechanism of action of trastuzumab.[5] Natural killer (NK) cells that express the Fc-gamma receptor are able to bind to trastuzumab and induce cell lysis. Trastuzumab was able to inhibit the outgrowth of macroscopically detectable xenograft tumors for up to 7 weeks in mice inoculated with positive and intrinsically trastuzumab-resistant HER2 cells. This effect is likely to be mediated via antibody-dependent cellular cytotoxicity, since the same phenomenon did not occur when trastuzumab-F(ab′) was used.[9]
Gennari et al evaluated 11 patients with HER2-positive breast cancer treated in the neoadjuvant setting with trastuzumab. Surgical samples, tumor biopsies, and lymphocytes from these patients were collected for biologic studies. Patients with complete or partial remissions were found to have a higher in situ infiltration of leukocytes and a higher capability to mediate in vitro antibody-dependent cellular cytotoxicity activity.[10]
Trastuzumab might also play a role as an antiangiogenic agent, since it has been shown to induce normalization and regression of the vasculature in a murine experimental human breast tumor model that overexpresses HER2.[11] The combined use of trastuzumab plus paclitaxel more effectively inhibited HER2-mediated angiogenesis than either treatment alone, resulting in more pronounced tumoricidal effects.
When overexpressed, the HER2 receptor is subjected to proteolysis, with the consequent cleavage and release to the circulation of the extracellular portion (p95HER2). The HER2 extracellular domain represents a truncated form of the HER2 receptor possessing intrinsic kinase activity in vitro. In patients with breast cancer, the presence of soluble extracellular domain appears to help distinguish tumors that have metastasized to the lymph nodes from those with negative nodes.[12] In addition, elevated levels of circulating HER2 extracellular domain in patients with metastatic breast cancer correlate with reduced efficacy of a paclitaxel/doxorubicin chemotherapy combination.[13] Trastuzumab treatment inhibits the proteolytic cleavage of HER2 and prevents the production of the active truncated HER2 fragment, which may constitute another mechanism of action of trastuzumab.[14]
Postulated Mechanisms of Resistance to Trastuzumab
Loss of PTEN
Patients with PTEN-deficient breast cancer had significantly poorer responses to trastuzumab-based therapy than those with normal PTEN.[8,15] Thus, PTEN deficiency might be a useful predictor of clinical resistance to trastuzumab.
Nagata et al have demonstrated that reducing PTEN in breast cancer cell lines by using antisense oligonucleotides conferred trastuzumab resistance in vitro and in vivo.[8] This process seems to be secondary to the reduction in AKT phosphorylation. Treatment of these tumor cells with inhibitors of the PI3K pathway was capable of overcoming resistance to trastuzumab. In that study, the analysis of 47 patients with metastatic breast cancer treated with trastuzumab plus a taxane showed that loss of PTEN expression was associated with lower response.[8]
Fujita et al have found a remarkable activity for trastuzumab in patients whose tumors had elevated PTEN expression on immunohistochemistry. Moreover, they found that duplex siRNA targeting PTEN significantly decreased the trastuzumab chemosensitivity of SKBR3 cells.[15]
These results suggest that the loss of PTEN can be used as a predictor of resistance to trastuzumab and that the use of drugs that inhibit PI3K can provide a therapeutic alternative in patients with low PTEN concentration.
Activation of Alternative Pathways
The insulin-like growth factor–I receptor (IGF-IR) is a transmembrane tyrosine kinase receptor associated with cell proliferation and metastasis formation. In breast cancer cell lines that overexpress HER2, an increased level of IGF-IR activation appears to interfere with the effectiveness of trastuzumab. Thus, strategies that target the IGF-IR signaling pathway may prevent or delay development of resistance to trastuzumab.[16]
Nahta et al observed a cross-talk between HER2 and IGF-IR in trastuzumab-resistant, but not in trastuzumab-sensitive, cell lines. The cross-talk between IGF-IR and HER2 in resistant cells is evidenced by the fact that IGF-I stimulation results in increased phosphorylation of HER2 in such cells; conversely, inhibition of IGF-IR tyrosine kinase activity leads to decreased HER2 phosphorylation in such resistant cells.[17]
The cyclin-dependent kinase inhibitor p27Kip1 was found to be decreased in trastuzumab-resistant breast tumor cells, and cyclin-dependent kinase 2 activity was increased. Importantly, the exogenous addition of p27Kip1 increased trastuzumab sensitivity. Additionally, resistant cells displayed heightened sensitivity to the proteasome inhibitor MG132, which induced p27Kip1 expression. Thus, trastuzumab resistance may be associated with decreased p27Kip1 levels and may be susceptible to treatments that induce p27Kip1 expression.[18]
These data suggest that reduction of the activity of p27Kip1 is associated with resistance to trastuzumab, possibly mediated by the heterodimerization of IGF-IR with HER2. Therefore, IGF-IR can be seen as an important potential therapeutic target in patients who are resistant to trastuzumab.
Receptor-Antibody Interaction Block
Another potential mechanism related to resistance to antibody-based therapy is the development of alterations that ultimately interfere with the interaction between the therapeutic agent and its target. The expression of MUC4, a membrane-associated mucin, seems to contribute to trastuzumab resistance by masking the HER2 receptor epitope. Overexpression of MUC4 has been shown to block cell-cell and cell-matrix interactions, protect tumor cells from immune surveillance, and promote metastasis. The expression of MUC4 was found to be higher in trastuzumab-resistant cell lines than in sensitive ones, and its level was inversely correlated with the trastuzumab binding capacity of single cells.
Abrogation of MUC4 expression by RNA interference increased the binding of trastuzumab, as described by Nagy et al[19] In MCF7 cell lines, the induction of MUC4 hyperexpression repressed the interaction between HER2 and HER2 antibodies, including trastuzumab.[20] In addition, MUC4 is being proposed as a ligand for HER2, and it also seems to increase the phosphorylation of HER2 and alter the signals generated from this receptor, without interfering with the expression of HER2.[19]
Although HER2 does not have a known direct ligand, HER2 heterodimerization with other receptors can be induced by ligands of EGFR, HER3, and HER4. In the presence of an excess of ligands, HER2, or both, the resulting heterodimers drive the activation of intracellular signaling using MAPK and PI3K pathways. It has been suggested that ligand overload can promote cell proliferation and diminish the effectiveness of trastuzumab.
Transforming growth factor (TGF)-alpha might play an important role in this resistance mechanism. The analysis of tumor samples from three breast cancer patients, performed before treatment with trastuzumab and after disease progression while still on therapy with the antibody, demonstrated the presence of TGF-alpha only after therapeutic failure. Moreover, in vitro expression of exogenous TGF-alpha in breast cancer cell lines was associated with a dramatic reduction in trastuzumab-induced HER2 endocytosis and cell growth inhibition.[21]
Analysis of the genetic material of trastuzumab-resistant cells disclosed the presence of an increased expression of the EGFR family binding factors, such as heregulin, TGF-alpha, and epidermal growth factor,[22] and also an increase in the dimerization between HER2/HER3 and HER2/EGFR.[23] These data suggest that the multiple and simultaneous blockade of different receptors of the EGFR family can be potentially more effective than the isolated blockade of HER2 by trastuzumab.
Finally, HER2 gene mutations can potentially alter the interaction between the receptor and trastuzumab. Although no available studies prove such a hypothesis, the presence of somatic mutations in the region that codes for the tyrosine kinase domain has already been described in some patients.[24]
HER2 Extracellular Domain


In a pooled analysis of seven trials of first-line trastuzumab therapy (with or without chemotherapy), a reduction of 20% or more in the serum concentration of HER2 extracellular domain was associated with increased progression-free and overall survival, compared with patients in whom a lesser reduction was observed.[25] In addition, based on an in vitro study using HER2-overexpressing breast tumor cells, Zabrecky et al suggested that extracellular domain can link to antibodies against HER2, thereby competing with the activated transmembrane HER2 receptor for the opportunity to interact with the antibody. This indicates another possible mechanism of resistance to treatment based on monoclonal antibodies against HER2.[26]
Possible TherapeuticStrategies to Overcome Trastuzumab Resistance
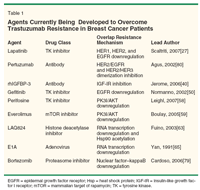
Given the recently reported mechanisms of trastuzumab resistance discussed previously, several novel therapies have the potential to be used with success in patients with primary or acquired resistance. The following discussion presents the available evidence on the most promising novel strategies, and some of the agents currently under development are shown in Table 1.
Inhibition of HER2 Tyrosine Kinase Activation
Lapatinib (Tykerb) is a small molecule administred orally that acts as a dual reversible inhibitor of the tyrosine kinase activity of EGFR and HER2, with a longer dissociation time and a more durable blockade than other tyrosine kinase inhibitors. As previously described, p95HER2 represents an altered form of the HER2 receptor derived from the cleavage of HER2 maintaining its tyrosine kinase activity, but which lacks the extracellular domain of the receptor and consequently no longer binds to trastuzumab. In vitro, the use of lapatinib is able to inhibit phosphorylation of p95HER2 and AKT, with consequent inhibition of cell growth.[27] The combination of lapatinib plus trastuzumab was found to markedly enhance tumor cell apoptosis in HER2-overexpressing breast cancer cells.[28]
A phase I/II study involving metastatic breast cancer patients tested the combination of lapatinib and trastuzumab, achieving a response rate of 22% and a stable disease rate of 37% in a heavily pretreated population.[29] Two nonrandomized multicenter studies evaluated the use of lapatinib in patients with HER2-positive metastatic breast cancer refractory to trastuzumab. The response rates (partial response, complete response, and stable disease) in the two studies were 14% and 22%, respectively.[30]
Spector and coworkers evaluated the use of lapatinib in patients with recurrent or refractory inflammatory, HER2-positive breast cancer, most of whom were already refractory to trastuzumab, showing a 62% partial response rate.[31] The role of lapatinib also was evaluated in HER2-positive breast cancer patients who had developed brain metastasis while on trastuzumab therapy. The response rate among these patients was 5%, and stable disease was achieved in another 20% of the women.[32]
More recently, Geyer et al published the first phase III study in HER2-positive metastatic breast cancer carried out in women who had been previously treated with a taxane, an anthracycline, and trastuzumab. Patients were randomized to receive capecitabine (Xeloda) with or without lapatinib. The study included 321 patients and was closed sooner than initially planned on the basis of the level of efficacy observed in the experimental arm. The interim analysis showed a 51% reduction in the risk of progression with the use of lapatinib. The combination group also reached a response rate of 23%, compared with 14% for single-agent capecitabine. The median time to progression was 8.4 months in the combination-therapy group, compared to 4.4 months in the monotherapy group. The study also showed a non–statistically significant reduction in the development of brain metastasis. These improvements were achieved without an increase in serious toxic effects or symptomatic cardiac events.[33]
Other Monoclonal Antibodies Against HER2
Pertuzumab (2C4) is an investigational monoclonal antibody that disrupts the dimerization between HER2 and its partners, EGFR and HER3, in both low– and high–HER2-expressing cells. It is also capable of disrupting the interaction between HER2 and IGF-IR in trastuzumab-resistant cells.[17]
Pertuzumab binds to extracellular domains of HER2 (domains I, II, and III), which differ from the trastuzumab binding site (domain IV).[34] The combination of the two antibodies was able to inhibit tumor growth and promote apoptosis[35] in previously untreated, HER2-overexpressing cells, suggesting that pertuzumab might be active in trastuzumab-resistant cells. When administered as a single agent, however, pertuzumab was unable to inhibit tumor growth in trastuzumab-resistant tumor cells,[36] probably because of the intracellular signaling alterations that were present in these cells.
The combination of pertuzumab, trastuzumab, and the EGFR tyrosine kinase inhibitor gefitinib (Iressa) was tested in an animal model. The simultaneous use of these three agents was able to inhibit tumor growth of HER2-overexpressing xenografts more effectively than the use of any of these drugs used as single agents or in dual combinations.[37]
Inhibition of the IGF-IR Pathway
As previously mentioned, activation of IGF-IR is one of the putative mechanisms leading to trastuzumab resistance. In vitro studies have shown that simultaneous cotargeting of the tyrosine kinase domains of HER2 and IGF-IR have a synergistic effect in terms of cell growth inhibition.[38] Triple blockade, using an antagonist of the estrogen receptor, HER2, and IGF-IR in cells that overexpress HER2 or that exhibit high levels of IGF-IR had a stronger apoptotic effect, compared with the simple or double blockade.[39]
A recently published study by Jerome and coworkers emphasizes the importance of IGF-IR blockade on the reversion of trastuzumab resistance. The authors studied a recombinant human IGF binding protein 3 (rhIGFBP-3), an antagonist of the IGF-IR signaling pathway, in trastuzumab-resistant breast cancer cells in vitro and in tumors in vivo. Its use in combination with trastuzumab in trastuzumab-resistant cancer cells was able to inhibit cell growth by means of the restoration of the intracellular downregulation of AKT and MAPK phosphorylation in vitro and in vivo.[40] Interestingly, lapatinib also inhibits IGF-I signaling and growth-promoting effects, enhancing the downregulation of tumor growth in trastuzumab-resistant cells when combined with rhIGFBP-3.[41]
Several monoclonal antibodies against IGF-IR are currently in development. The investigational agent CP-751,871[42] is a fully human IgG2 antibody with high affinity for human IGF-IR, already in evaluation in phase II studies in association with carboplatin and paclitaxel in non–small-cell lung cancer.[43-45] IMC-A12 is a fully human monoclonal IgG1 antibody. This agent has great affinity for IGF-IR and, in athymic mice, presents synergistic activity with EGFR inhibitors.
AMG479 is a fully human monoclonal antibody that binds to IGF-I and IGF-II receptors. The results of the phase I study reported at the American Society of Clinical Oncology (ASCO) annual meeting in 2007 have established reversible grade 3/4 thrombocytopenia as the main adverse effect of AMG479. Objective responses (partial and complete) were observed in Ewing’s sarcoma and carcinoid tumors.[46]
R1507 is a human monoclonal antibody directed against the IGF-1R. In phase I trials, frequent stabilization of the disease was observed.[47] Finally, the selective inhibitor of IGF-IR kinase, NVP-AEW541,[48] is a new targeted agent against IGF-IR that is currently being tested in trastuzumab-resistant breast cancer cell lines.[49]
Inhibition of the EGFR Pathway
Preclinical studies suggested that trastuzumab and gefitinib might have a potential synergistic effect on the inhibition of cell growth.[50,51] However, a phase II study lead by Arteaga in women with advanced HER2-positive breast cancer treated with trastuzumab plus gefitinib was prematurely closed due to low effectiveness of the combination, suggesting potential antagonism between these two agents.[52]
According to an evaluation of 90 patients with stage II/IIIA breast cancer treated primarily with the combination of epirubicin and paclitaxel, with or without the addition of gefitinib, the use of gefitinib plus chemotherapy did not result in meaningful effects on the EGFR-dependent pathway, proliferation, apoptosis, or expression of the vascular endothelial growth factor receptor 2, as compared with placebo, but did produce increased skin and mucosal toxicity.[53] The same negative results were found in a study involving 216 postmenopausal patients with hormone-receptor–positive tumors when anastrozole (Arimidex) was used in the neoadjuvant setting in combination with gefitinib.[54] However, a similar study involving 56 patients found a synergistic effect between gefitinib and anastrozole.[55] A variety of mechanisms have been postulated to explain the resistance of breast cancer cells to gefitinib, given that its action does not depend on the presence of the EGFR mutation, as in lung cancer.
CI-1033, a pan-ErbB tyrosine kinase inhibitor, is a clinically promising agent that is active against all four members of the ErbB receptor tyrosine kinase family (HER1, HER2, HER3, and HER4). In vitro studies of human cancer cell lines indicate that CI-1033 results in prompt, potent, and sustained inhibition of tyrosine kinase activity.[56] Animal studies showed some activity on different tumors types. The therapeutic effect of this agent was related to downregulation of the EGFR and HER2 phosphorylation, which was observed in 40% to 50% of cases, based on the evaluation of posttreatment biopsies.[57] Clinical studies are currently evaluating CI-1033 in patients with trastuzumab-resistant, HER2-positive breast cancer.
Inhibition of PI3K
Inhibition of the PK13/AKT pathway is being evaluated as a potential way to overcome trastuzumab resistance. Perifosine is an inhibitor of the AKT pathway that was recently evaluated in a phase II study involving 17 patients with previously treated metastatic breast cancer. Sadly, no objective responses were seen in this group of patients.[58] Another mechanism of blockading the PI3K-signaling pathway is the inhibition of the mammalian target of rapamycin (mTOR). In hormone-sensitive breast cancer cells, the combination of the mTOR inhibitor everolimus (RAD001) with letrozole (Femara) had a synergistic effect on cell growth inhibition.[59] The use of the mTOR inhibitor temsirolimus (Torisel) as a single agent produced an objective response rate of 9.2% (partial responses), and the median time to tumor progression was 12 weeks.[60]
Lu et al have conducted preclinical studies to access the efficacy and clinical applicability of inhibitors of the AKT/mTOR pathways as a way to restore trastuzumab sensitivity to PTEN-deficient cells.[61] Only two of six drugs evaluated were able to overcome trastuzumab resistance. When trastuzumab was combined with the AKT inhibitor triciribine, breast cancer cell growth was inhibited and apoptosis was induced. In a xenograft model, the combination of trastuzumab and triciribine dramatically inhibited tumor growth. The combination of trastuzumab and the mTOR inhibitor everolimus also slowed breast cancer cell growth in vitro and in vivo. Such data show the potential of these drug combinations as alternatives for overcoming trastuzumab resistance.
Inhibition of Histone Deacetylation and the Use of Heat Shock Protein
The chaperone heat shock protein (Hsp)90, besides catalyzing the proper folding of newly synthesized client proteins into a stable tertiary conformation, has been implicated in the stabilization of a number of cellular proteins that play central roles in the signal transduction processes, including HER2. Interestingly, binding of Hsp90 to HER2 not only serves to maintain its physiologic conformation, but also to restrain HER2 from forming active signaling dimers. The inhibition of Hsp90 results in the proteasomal degradation of its maintained proteins (eg, HER2, AKT, and Raf-1) and, as a result, apoptosis of the target cell. Geldanamycin and its derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG) are inhibitors of Hsp90, and 17-AAG that has the same capacity to induce apoptosis in trastuzumab-resistant breast tumor cells when combined with the antibody.[62] Clinical studies are evaluating its efficacy in trastuzumab-resistant breast cancer patients.
Histone deacetylase inhibitors induce hyperacetylation of the amino-terminal lysine residues of the core nucleosomal histones, which results in chromatin remodeling and altered gene expression. Recent studies demonstrate that the exposure to a novel hydroxamic acid analog histone deacetylase inhibitor, LAQ824, induced p21Waf1 and p27Kip1 and caused cell growth arrest and apoptosis of human breast cancer cells that possess amplification and overexpression of HER2. Treatment with LAQ824 depleted the mRNA and protein levels of HER2, which, in turn, was associated with attenuation of phosphorylated AKT, c-Raf-1, and phosphorylated MAPK levels. LAQ824 also induced the acetylation of Hsp90, resulting in inhibition of its binding to ATP, which has been shown to impair the chaperone association of Hsp 90 with its client proteins, HER2, AKT, and c-Raf. LAQ824 also enhanced docetaxel (Taxotere)-induced, epothilone B–induced, and gemcitabine (Gemzar)-induced apoptosis of HER2-positive breast cancer cells.[63]
Suberoylanilide hydroxamic acid (SAHA) is another inductor of the acetylation of Hsp90 capable of inducing apoptosis in breast cancer cells with HER2 amplification and improving the apoptotic effects of trastuzumab and docetaxel.[64] These data suggest that it might be worthwhile to design clinical studies with histone deacetylase inhibitors in patients with HER2-positive breast cancer.
Gene Therapy
Yan et al reported that the HER2 gene product p185 can be downregulated by the adenovirus E1A gene, which is responsible for the repression of HER2 at the RNA level.[65] The delivery of the E1A gene by both adenovirus-mediated transfection and by cationic liposomes in tumors with HER2 overexpression induced the inhibition of tumor growth and prolongation of survival in animal models.[66] In vitro, the adenovirus type 5 E1A gene can sensitize paclitaxel-resistant, HER2-overexpressing breast cancer cells to the drug by repressing HER2 expression.[67] Such data provided the rationale for a phase I study that was conducted by Yoo et al in 18 patients with unresectable breast or head and neck cancer. In this study, no limiting toxicity was recorded when liposomes containing the gene E1A were injected in these patients,[68] allowing for further development of clinical trials.
Vaccines



Vaccines have the ability to stimulate or activate the immune system through the activation of cytokines, proteins, or growth factors. Recently, several types of vaccines (peptides, DNA, and proteins) have been developed against the HER2 receptor. Vaccination with the peptide E75 associated with granulocyte-macrophage colony-stimulating factor (GM-CSF, Leukine) was tested in 14 patients with metastatic breast or ovarian cancer and was well tolerated, inducing the generation of protein-specific and epitope-specific immunity.[69] In another study, 92% percent of patients developed T-cell immunity to HER2 peptides and 68% to a HER2 protein domain. At 1-year follow-up, immunity to the HER2 protein persisted in 38% of patients.[70]
A clinical study involving 53 lymph node–positive breast cancer patients, with no evidence of disease demonstrated that the vaccine against HER2 (E75) produced clonal expansion of E75-specific, CD8-positive T cells that lysed HER2-expressing tumor cells in vitro. This strategy showed an acceptable safety profile and a trend toward a reduction of the tumor recurrence rate.[71]
The HER2 intracellular domain protein vaccine was also effective in eliciting HER2-specific T-cell and antibody immunity in the majority (89%) of breast and ovarian cancer patients treated with the regimen. More than 50% of cases retained specific immunity between 9 and 12 months after the end of treatment.[72] Treatment of breast tumor cells with trastuzumab promotes a toxicity mediated by lymphocytes that is independent of the expression of HER2, and this effect is improved with the use of these vaccines.[73]
Another type of vaccine using a murine monoclonal anti-Id antibody, 6D12, which mimics a specific epitope of HER2, can be used as a surrogate antigen for HER2 monoclonal antibodies.[74] Peptides constructed to mimic the dimerization region of HER2 receptor have also been used in vaccine development. In vitro and in vivo studies showed that these molecules generate a strong stimulation in the production of specific antibodies that inhibited the intracellular signaling of HER2, leading to an antibody-mediated cellular toxicity and, thus, reduction of tumor development.[75]
Lapuleucel-T (APC8024), an autologous active cellular immunotherapy, is prepared from peripheral blood mononuclear cells, including antigen-presenting cells, which were activated in vitro with recombinant fusion antigens. This antigen construct consisted of sequences from intracellular and extracellular domains of HER2 linked to GM-CSF. A phase I study involving 18 patients with metastatic HER2-positive breast cancer was conducted. The treatment resulted in significant immune responses (one partial response and six cases of stable disease), which were enhanced after boost infusions without evidence of the development of any significant toxicities.[76]
Finally, 2B1 is a bispecific murine monoclonal antibody that binds to HER2 extracellular domain and Fc-gammaRIII and that efficiently promotes the lysis of tumor cells overexpressing HER2 by NK cells and mononuclear phagocytes that express the Fc-gammaRIII A isoform. A phase Ib/II study led by the Eastern Cooperative Oncology Group demonstrated the ability of this antibody to induce detectable immune responses against intracellular and extracellular domains of HER2 in some of the 20 women with metastatic breast cancer who were exposed to this agent. Nevertheless, this immunity was not enough to produce any meaningful clinical responses.[77]
Other Mechanisms
ScFv(FRP5)-ETA is a recombinant antibody toxin with binding specificity for HER2. It consists of an N-terminal single-chain antibody fragment (scFv), genetically linked to truncated Pseudomonas exotoxin A (ETA). Potent antitumoral activity of scFv(FRP5)-ETA against HER2-overexpressing tumor cells was demonstrated in vitro and in animal models. Azemar et al treated 11 patients with metastatic malignant melanoma, breast cancer, and colorectal cancer with intratumoral injections of scFv(FRP5)-ETA into cutaneous lesions once daily for 7 to 10 days. Treatment caused injected tumors to shrink in 6 of the 10 cases evaluated. Complete regression of injected tumor nodules was accomplished in four patients, and partial reduction in tumor size in another two patients. Adverse reactions were restricted to local symptoms such as pain and inflammation at injection sites, which were fully reversible.[78]
The proteasome is a large, multi-subunit protein enzymatic complex responsible for the degradation of nuclear proteins. It is present in high amounts both in the cytoplasm and in the nucleus, and is responsible for eliminating cellular proteins, including proteins that have been tagged for degradation through polyubiquitination. Proteins entering the proteasome are degraded through catalytic activities that take place within the core of the proteasome. Substrates for degradation include a variety of proteins that are responsible for critical functions such as regulation of the cell cycle, transcription, and apoptosis, as well as the regulation of chemotaxis, angiogenesis, and cell adhesion. Bortezomib (Velcade), also known as PS-341, was the first proteasome inhibitor to be approved for clinical use, and is active in patients with refractory multiple myeloma.
Overexpression of HER2 in mammary cells leads to AKT phosphorylation and consequent increase in nuclear factor–kappaB (NF-κB) expression. The nuclear translocation of NF-κB promotes the activation of different antiapoptotic and prometastatic genes, as well as proangiogenic factors. HER2 also promotes the proteasomic degradation of p27Kip1 with subsequent cell-cycle activation. Therefore bortezomib is able to increase p27Kip1 expression and to reduce the nuclear concentration of NF-κB, possibly increasing the effect of trastuzumab. In one in vitro analysis using mammary cells with different levels of HER2 expression, the combination of trastuzumab and bortezomib had a synergistic effect in cell-death induction, and this effect was related to an increase in NF-κB expression and low nuclear expression of p27Kip1.[79] Phase I trials evaluating this drug combination are in progress.
Discussion and Conclusion
The recognition of the HER2-overexpressing breast cancer phenotype as a clinical entity was one of most important lessons learned in medical oncology during the latter part of the 20th century. The knowledge acquired through the study of HER2-overexpressing tumors led to the clinical development of trastuzumab, the first monoclonal antibody to be approved for the treatment of a solid tumor. In this review, we tried to describe the several different mechanisms involved in resistance to trastuzumab, as well as to describe the potential therapeutic opportunities related to each specific resistance mechanism, with examples of agents that are currently being developed.
Today, the determination of HER2 status constitutes an indispensable part of the initial tumor evaluation of each breast cancer patient. However, even though the use of trastuzumab is based on the presence of pathologic HER2 overexpression (as determined by either immunohistochemistry or fluorescence in situ hybridization), the simple presence of HER2 is neither sufficient to ensure that trastuzumab will be effective in first-line treatment nor a guarantee that acquired resistance will not develop. We do not currently understand the underlying causes of treatment failure in every patient. As a consequence, treatment strategies employed in the treatment of HER2-resistant tumors-such as the continuation of trastuzumab treatment using a different chemotherapy combination or the introduction of lapatinib in association with capecitabine-are rather empiric.
Therefore, understanding the underlying mechanisms of action of trastuzumab becomes critical so that we can develop strategies to prevent or overcome resistance by accounting for the mechanisms involved in treatment failure. Ideally, we hope that in the future we may establish the specific basis of treatment failure in each patient, in order to use our targeted agents more rationally.
References:
References
1. Slamon DJ, Godolphin W, Jones LA, et al: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244:707-712, 1989.
2. Slamon DJ, Leyland-Jones B, Shak S, et al: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783-792, 2001.
3. Viani GA, Afonso SL, Stefano EJ, et al: Adjuvant trastuzumab in the treatment of her-2-positive early breast cancer: A meta-analysis of published randomized trials. BMC Cancer 7:153, 2007.
4. Yarden Y, Sliwkowski MX: Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127-137, 2001.
5. Baselga J, Albanell J, Molina MA, et al: Mechanism of action of trastuzumab and scientific update. Semin Oncol 28:4-11, 2001.
6. Delord JP, Allal C, Canal M, et al: Selective inhibition of HER2 inhibits AKT signal transduction and prolongs disease-free survival in a micrometastasis model of ovarian carcinoma. Ann Oncol 16:1889-1897, 2005.
7. Lane HA, Motoyama AB, Beuvink I, et al: Modulation of p27/Cdk2 complex formation through 4D5-mediated inhibition of HER2 receptor signaling. Ann Oncol 12(suppl 1):S21-S2, 2001.
8. Nagata Y, Lan KH, Zhou X, et al: PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6:117-127, 2004.
9. Barok M, Isola J, Palyi-Krekk Z, et al: Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol Cancer Ther 6:2065-2072, 2007.
10. Gennari R, Menard S, Fagnoni F, et al: Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clin Cancer Res 10:5650-5655, 2004.
11. Izumi Y, Xu L, di Tomaso E, et al: Tumour biology: Herceptin acts as an anti-angiogenic cocktail. Nature 416:279-280, 2002.
12. Christianson TA, Doherty JK, Lin YJ, et al: NH2-terminally truncated HER-2/neu protein: Relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Res 58:5123-5129, 1998.
13. Colomer R, Montero S, Lluch A, et al: Circulating HER2 extracellular domain and resistance to chemotherapy in advanced breast cancer. Clin Cancer Res 6:2356-2362, 2000.
14. Molina MA, Codony-Servat J, Albanell J, et al: Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res 61:4744-4749, 2001.
15. Fujita T, Doihara H, Kawasaki K, et al: PTEN activity could be a predictive marker of trastuzumab efficacy in the treatment of ErbB2-overexpressing breast cancer. Br J Cancer 94:247-252, 2006.
16. Lu Y, Zi X, Zhao Y, et al: Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst 93:1852-1857, 2001.
17. Nahta R, Yuan LX, Zhang B, et al: Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 65:11118-11128, 2005.
18. Nahta R, Takahashi T, Ueno NT, et al: P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res 64:3981-3986, 2004.
19. Nagy P, Friedlander E, Tanner M, et al: Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res 65:473-482, 2005.
20. Price-Schiavi SA, Jepson S, Li P, et al: Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer 99:783-791, 2002.
21. Valabrega G, Montemurro F, Sarotto I, et al: TGFalpha expression impairs Trastuzumab-induced HER2 downregulation. Oncogene 24:3002-3010, 2005.
22. Ritter CA, Perez-Torres M, Rinehart C, et al: Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res 13:4909-4919, 2007.
23. Diermeier S, Horvath G, Knuechel-Clarke R, et al: Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp Cell Res 304:604-619, 2005.
24. Lee JW, Soung YH, Seo SH, et al: Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res 12:57-61, 2006.
25. Ali SM, Esteva FJ, Fornier M, et al: Serum HER-2/neu change predicts clinical outcome to trastuzumab-based therapy (abstract 500). J Clin Oncol 24(18S):3s, 2006.
26. Zabrecky JR, Lam T, McKenzie SJ, et al: The extracellular domain of p185/neu is released from the surface of human breast carcinoma cells, SK-BR-3. J Biol Chem 266:1716-1720, 1991.
27. Scaltriti M, Rojo F, Ocana A, et al: Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst 99:628-638, 2007.
28. Xia W, Gerard CM, Liu L, et al: Combining lapatinib (GW572016), a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases, with therapeutic anti-ErbB2 antibodies enhances apoptosis of ErbB2-overexpressing breast cancer cells. Oncogene 24:6213-6221, 2005.
29. Storniolo AM, Burris H, Pegram M, et al: A phase I, open-label study of lapatinib (GW572016) plus trastuzumab; a clinically active regimen (abstract 559). J Clin Oncol 23(16S):18s, 2005.
30. Blackwell KL, Burstein H, Pegram M, et al: Determining relevant biomarkers from tissue and serum that may predict response to single agent lapatinib in trastuzumab refractory metastatic breast cancer (abstract 3004). J Clin Oncol 23(16S):193s, 2005.
31. Spector NL, Blackwell K, Hurley J, et al: EGF103009, a phase II trial of lapatinib monotherapy in patients with relapsed/refractory inflammatory breast cancer (IBC): Clinical activity and biologic predictors of response (abstract 502). J Clin Oncol 24(18S):3s, 2006.
32. Lin NU, Carey LA, Liu MC, et al: Phase II trial of lapatinib for brain metastases in patients with HER2+ breast cancer (abstract 503). J Clin Oncol 24(18S):3s, 2006.
33. Geyer CE, Forster J, Lindquist D, et al: Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733-2743, 2006.
34. Cho HS, Mason K, Ramyar KX, et al: Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421:756-760, 2003.
35. Nahta R, Hung MC, Esteva FJ: The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res 64:2343-2346, 2004.
36. Tanner M, Kapanen AI, Junttila T, et al: Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol Cancer Ther 3:1585-1592, 2004.
37. Arpino G, Gutierrez C, Weiss H, et al: Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J Natl Cancer Inst 99:694-705, 2007.
38. Camirand A, Lu Y, Pollak M: Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit 8:BR521-BR526, 2002.
39. DiGiovanna MP, Chakraborty A: Combinations of HER2, estrogen receptor (ER) and IGF-I receptor (IGF1R) inhibitors induce apoptosis in breast cancer cells: Dramatic effects of HER2 inhibitors on non-overexpressing cells (abstract 1226). Proc Am Assoc Cancer Res 47, 2006.
40. Jerome L, Alami N, Belanger S, et al: Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res 66:7245-7252, 2006.
41. Nahta R, Yuan LX, Du Y, et al: Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: Effects on insulin-like growth factor I signaling. Mol Cancer Ther 6:667-674, 2007.
42. Cohen BD, Baker DA, Soderstrom C, et al: Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res 11:2063-2073, 2005.
43. Lacy M, Alsina M, Melvin CL, et al: Phase 1 first-in-human dose escalation study of cp-751,871, a specific monoclonal antibody against the insulin like growth factor 1 receptor (abstract 7609). J Clin Oncol 24(18S):448s, 2006.
44. Pollak MN, Lacy MQ, Lipton A, et al: Pharmacodynamic properties of the anti-IGF-IR monoclonal antibody CP-751,871 in cancer patients (abstract 3587). J Clin Oncol 25(18S):159s, 2007.
45. Haluska P, Shaw H, Batzel GN, et al: Phase I dose escalation study of the anti-IGF-1R monoclonal antibody CP-751,871 in patients with refractory solid tumors (abstract 3586). J Clin Oncol 25(18S):159s, 2007.
46. Tolcher AW, Rothenberg ML, Rodon J, et al: A phase I pharmacokinetic and pharmacodynamic study of AMG 479, a fully human monoclonal antibody against insulin-like growth factor type 1 receptor (IGF-1R), in advanced solid tumors (abstract 3002). J Clin Oncol 25(18S):118s, 2007.
47. Rodon J, Patnaik A, Stein M, et al: A phase I study of q3W R1507, a human monoclonal antibody IGF-1R antagonist in patients with advanced cancer (abstract 3590). J Clin Oncol 25(18S):160s, 2007.
48. Garcia-Echeverria C, Pearson MA, Marti A, et al: In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 5:231-239, 2004.
49. Esparis-Ogando A, Rodriguez-Barrueco R, Borges J, et al: Insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 is active in breast cancer cells and enhances growth inhibition by herceptin through an increase in cell cycle arrest (abstract 21077). J Clin Oncol 25(18S):735s, 2007.
50. Normanno N, Campiglio M, De LA, et al: Cooperative inhibitory effect of ZD1839 (Iressa) in combination with trastuzumab (Herceptin) on human breast cancer cell growth. Ann Oncol 13:65-72, 2002.
51. Warburton C, Dragowska WH, Gelmon K, et al: Treatment of HER-2/neu overexpressing breast cancer xenograft models with trastuzumab (Herceptin) and gefitinib (ZD1839): Drug combination effects on tumor growth, HER-2/neu and epidermal growth factor receptor expression, and viable hypoxic cell fraction. Clin Cancer Res 10:2512-2524, 2004.
52. Moulder SL, Arteaga CL: A phase I/II trial of trastuzumab and gefitinib in patients with metastatic breast cancer that overexpresses HER2/neu (ErbB-2). Clin Breast Cancer 4:142-145, 2003.
53. Guarneri V, Frassoldati A, Ficarra G, et al: Phase II, randomized trial of preoperative epirubicin-paclitaxel +/- gefitinib with biomarker evaluation in operable breast cancer. Breast Cancer Res Treat 110:127-134, 2007.
54. Smith IE, Walsh G, Skene A, et al: A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol 25:3816-3822, 2007.
55. Polychronis A, Sinnett HD, Hadjiminas D, et al: Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: A double-blind placebo-controlled phase II randomised trial. Lancet Oncol 6:383-391, 2005.
56. Rusnak DW, Affleck K, Cockerill SG, et al: The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: potential therapy for cancer. Cancer Res 61:7196-7203, 2001.
57. Slichenmyer WJ, Elliott WL, Fry DW: CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin Oncol 28:80-85, 2001.
58. Leighl NB, Dent S, Clemons M, et al: A phase 2 study of perifosine in advanced or metastatic breast cancer. Breast Cancer Res Treat 108:87-92, 2007.
59. Boulay A, Rudloff J, Ye J, et al: Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 11:5319-5328, 2005.
60. Chan S, Scheulen ME, Johnston S, et al: Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23:5314-5322, 2005.
61. Lu CH, Wyszomierski SL, Tseng LM, et al: Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res 13:5883-5888, 2007.
62. Zsebik B, Citri A, Isola J, et al: Hsp90 inhibitor 17-AAG reduces ErbB2 levels and inhibits proliferation of the trastuzumab resistant breast tumor cell line JIMT-1. Immunol Lett 104:146-155, 2006.
63. Fuino L, Bali P, Wittmann S, et al: Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther 2:971-984, 2003.
64. Bali P, Pranpat M, Swaby R, et al: Activity of suberoylanilide hydroxamic acid against human breast cancer cells with amplification of her-2. Clin Cancer Res 11:6382-6389, 2005.
65. Yan DH, Chang LS, Hung MC: Repressed expression of the HER-2/c-erbB-2 proto-oncogene by the adenovirus E1a gene products. Oncogene 6:343-345, 1991.
66. Chang JY, Xia W, Shao R, et al: The tumor suppression activity of E1A in HER-2/neu-overexpressing breast cancer. Oncogene 14:561-568, 1997.
67. Ueno NT, Yu D, Hung MC: Chemosensitization of HER-2/neu-overexpressing human breast cancer cells to paclitaxel (Taxol) by adenovirus type 5 E1A. Oncogene 15:953-960, 1997.
68. Yoo GH, Hung MC, Lopez-Berestein G, et al: Phase I trial of intratumoral liposome E1A gene therapy in patients with recurrent breast and head and neck cancer. Clin Cancer Res 7:1237-1245, 2001.
69. Murray JL, Gillogly ME, Przepiorka D, et al: Toxicity, immunogenicity, and induction of E75-specific tumor-lytic CTLs by HER-2 peptide E75 (369-377) combined with granulocyte macrophage colony-stimulating factor in HLA-A2+ patients with metastatic breast and ovarian cancer. Clin Cancer Res 8:3407-3418, 2002.
70. Disis ML, Gooley TA, Rinn K, et al: Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. J Clin Oncol 20:2624-2632, 2002.
71. Peoples GE, Gurney JM, Hueman MT, et al: Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J Clin Oncol 23:7536-7545, 2005.
72. Disis ML, Schiffman K, Guthrie K, et al: Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein-based vaccine. J Clin Oncol 22:1916-1925, 2004.
73. Mittendorf EA, Storrer CE, Shriver CD, et al: Investigating the combination of trastuzumab and HER2/neu peptide vaccines for the treatment of breast cancer. Ann Surg Oncol 13:1085-1098, 2006.
74. Mohanty K, Saha A, Pal S, et al: Anti-tumor immunity induced by an anti-idiotype antibody mimicking human Her-2/neu. Breast Cancer Res Treat 104:1-11, 2007.
75. Allen SD, Garrett JT, Rawale SV, et al: Peptide vaccines of the HER-2/neu dimerization loop are effective in inhibiting mammary tumor growth in vivo. J Immunol 179:472-482, 2007.
76. Park JW, Melisko ME, Esserman LJ, et al: Treatment with autologous antigen-presenting cells activated with the HER-2 based antigen lapuleucel-T: results of a phase I study in immunologic and clinical activity in HER-2 overexpressing breast cancer. J Clin Oncol 25:3680-3687, 2007.
77. Borghaei H, Alpaugh RK, Bernardo P, et al: Induction of adaptive Anti-HER2/neu immune responses in a phase 1B/2 trial of 2B1 bispecific murine monoclonal antibody in metastatic breast cancer (E3194): a trial coordinated by the Eastern Cooperative Oncology Group. J Immunother 30:455-467, 2007.
78. Azemar M, Djahansouzi S, Jager E, et al: Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res Treat 82:155-164, 2003.
79. Cardoso F, Durbecq V, Laes JF, et al: Bortezomib (PS-341, Velcade) increases the efficacy of trastuzumab (Herceptin) in HER-2-positive breast cancer cells in a synergistic manner. Mol Cancer Ther 5:3042-3051, 2006.
80. Agus DB, Akita RW, Fox WD, et al: Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2:127-137, 2002.











