Emerging Role of EGFR-Targeted Therapies and Radiation in Head and Neck Cancer
The treatment of head and neck cancer has been at the forefront ofnovel therapeutic paradigms. The introduction of drugs that interactwith selective biologic pathways in the cancer cell has generated considerableattention recently. A wide variety of new compounds that attemptto target growth-signaling pathways have been introduced intothe clinic. A majority of studies in the clinic have focused on epidermalgrowth factor receptor (EGFR) antagonists, but future studies will likelybuild upon or complement this strategy with agents that target angiogenicor cell-cycle pathways. EGFR activation promotes a multitude ofimportant signaling pathways associated with cancer development andprogression, and importantly, resistance to radiation. Since radiationtherapy plays an integral role in managing head and neck squamouscell cancer (HNSCC), inhibiting the EGFR pathway might improveour efforts at cancer cure. The challenge now is to understand whenthe application of these EGFR inhibitors is relevant to an individualpatient and how or when these drugs should be combined with radiationor chemotherapy. Are there molecular markers available to determinewho will respond to EGFR inhibitors and who should be treatedwith alternative approaches? What are the mechanisms behind intrinsicor acquired resistance to targeted agents, and how do we preventthis problem? We need to formulate integrated laboratory/clinicalresearch programs that address these important issues.
The treatment of head and neck cancer has been at the forefront of novel therapeutic paradigms. The introduction of drugs that interact with selective biologic pathways in the cancer cell has generated considerable attention recently. A wide variety of new compounds that attempt to target growth-signaling pathways have been introduced into the clinic. A majority of studies in the clinic have focused on epidermal growth factor receptor (EGFR) antagonists, but future studies will likely build upon or complement this strategy with agents that target angiogenic or cell-cycle pathways. EGFR activation promotes a multitude of important signaling pathways associated with cancer development and progression, and importantly, resistance to radiation. Since radiation therapy plays an integral role in managing head and neck squamous cell cancer (HNSCC), inhibiting the EGFR pathway might improve our efforts at cancer cure. The challenge now is to understand when the application of these EGFR inhibitors is relevant to an individual patient and how or when these drugs should be combined with radiation or chemotherapy. Are there molecular markers available to determine who will respond to EGFR inhibitors and who should be treated with alternative approaches? What are the mechanisms behind intrinsic or acquired resistance to targeted agents, and how do we prevent this problem? We need to formulate integrated laboratory/clinical research programs that address these important issues.
Squamous cell carcinoma of the head and neck (HNSCC) accounts for greater than 90% of all upper aerodigestive tract malignancies and represents the sixth most common neoplasm in the world.[1,2] For these patients, survival outcome has not significantly improved in 25 years despite advances in surgery, radiation, and chemotherapy.[3,4] Part of the difficulty in treating HNSCC is that this designation actually represents a heterogeneous group of neoplasms arising from different anatomic subsites, the optimal therapy for which has yet to be defined. Current trends incorporate concurrent chemoradiotherapy; however, acute and late toxicities can be quite severe, increasing the chance of cure at the expense of quality of life. Shifting focus to strategies that identify certain genes and proteins associated with unregulated cancer cell growth and invasion common to HNSCC may allow us to refine our treatments for this disease, improve outcomes, and possibly reduce morbidity.
Growth factor receptors and their associated tyrosine kinases have been found to play key roles in oncogenesis and progression of HNSCC. Members of the erbB (also known as HER) family including erbB1-more commonly referred to as the epidermal growth factor receptor (EGFR)-have been intensively studied in the past decade. A variety of solid tumors, including HNSCC are known to express high levels of EGFR.[5] The prognostic-predictive value of EGFR expression in HNSCC has been shown in several studies including a correlative analysis of patients enrolled into a phase III trial conducted by the Radiation Therapy Oncology Group.[6]
In theory, the blockade of EGFR should result in the inhibition of tumor growth, and HNSCC was one of the first tumor models for which selective targeting of the EGFR in combination with radiation or chemotherapy were developed in the laboratory and moved to clinical testing. Novel therapeutic agents that target EGFR and its downstream signal pathway are currently being developed. These include monoclonal antibodies to EGFR, tyrosine kinase inhibitors, and agents that block EGFR transcription. In this paper, we will review the role of EGFR-mediated signal transduction in HNSCC development and examine various therapeutic strategies that target EGFR alone or in combination with radiation in the treatment of HNSCC.
ErbB Receptor Signaling in HNSCC
FIGURE 1


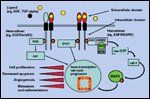
EGFR Biology
What role does the EGFR signaling pathway play in HNSCC, and is it relevant to outcomes? To begin with, EGFR is a member of the family of cell surface tyrosine kinase receptors that includes erbB receptors: EGFR/erbB1, erbB2/HER2-neu, erbB3/HER3, and erbB4/HER4. The EGF receptor is a 170-kD transmembrane glycoprotein whose gene is located on the short arm of chromosome 7 and consists of an extracellular binding domain, a transmembrane section, and an intracellular region possessing intrinsic tyrosine kinase activity (Figure 1).[7] Ligand binding by either EGF or transforming growth factor (TGF)-alpha induces the formation of homo- or heterodimeric complexes, with various family members of EGFR, in turn, activating intrinsic tyrosine kinases (Figure 1).
This autophosphorylation causes an activation of downstream pathways including Ras/MAP kinase, phosphatidylinositol- 3 kinase, and signal transducers and activators of transcription (STAT) proteins.[8] Transduction of these signal pathways has been associated with steps critical to carcinogenesis and cancer progression such as (1) the activation of redundant intracellular signaling pathways to promote cancer survival and prevent apoptosis, (2) cell proliferation, (3) angiogenesis and invasion, and (4) radioresistance.
Although EGFR has been the most widely studied of these receptors, it is notable that coexpression of erbB2, erbB3, and erbB4 have all been associated with decreased survival in patients with HNSCC.[9] ErbB2 and erbB3 overexpression is seen in 20% and 46% of oral cavity tumors, respectively, predicting poorer survival and the presence of nodal metastatic disease.[10,11] In some cancers (eg, gliomas), parts of the extracellular domain of EGFR are deleted or lost, resulting in constitutive activation of the receptor. In addition, aberrant production of EGF-related ligand promotes an autocrine loop for EGFR activation. The conclusive message is that many epithelial cancers overexpress multiple erbB receptors, which result in transformation and, later, proliferation and resistance of the cancer cell to therapeutic agents including radiation.
EGFR Expression in HNSCC
Compared with levels seen in the normal mucosa of patients without cancer, overexpression of EGFR or TGF-alpha has been reported in 80% to 90% of HNSCC tumors.[12] EGFR overexpression has been shown to be an independent prognostic factor in patients with HNSCC, and elevated EGFR protein levels are associated with decreased disease-free and causespecific survival. Furthermore, EGFR mRNA and protein are overexpressed in dysplastic lesions and in surrounding histologically normal mucosa of patients with HNSCC, suggesting that EGFR upregulation represents an early event in HNSCC carcinogenesis.[13]
In patients with HNSCC, field cancerization or the presence of a "condemned mucosa" is likely and may be related to the high levels of EGFR expressed in the histologically normal tissue. As to the cause of constitutive EGFR upregulation, the main mechanism appears to be transcriptional activation. This is primarily related to autocrine production of TGF-alpha, although EGFR gene amplification has been detected in some HNSCC cell lines.[14]
EGFR amplification and poor survival in patients with lung or head and neck cancer was first reported more than a decade ago.[15] A higher incidence of EGFR expression was noted in patients with T1/2 glottic and subglottic carcinomas who experienced recurrence.[16] Immunohistochemical (IHC) analysis of the expression of EGFR and of its ligand, TGF-alpha, in 91 patients with various stages (T1-4, N0/1) and sites of HNSCC all treated with surgical resection (and a proportion also treated with radiotherapy or chemotherapy) revealed that the combination of EGFR or TGF-alpha level with lymph node stage was the strongest predictor of cause-specific survival.[17]
FIGURE 2



EGFR Expression and Postirradiation Outcome in Patients With Head and Neck Cancer
Confirming these findings, Ang et al conducted an IHC analysis of tumor specimens of patients with locally advanced HNSCC treated in a Radiation Therapy Oncology Group study (RTOG 90-03) evaluating outcomes based on different radiation schedules.[6] Of the 155 specimens available for analysis, 95% had detectable EGFR expression. As shown in Figure 2, high (> median) EGFR expression was significantly correlated with poorer overall and lower disease-free survival rates in multivariate analyses (P = .003). Interestingly, no correlation was noted between EGFR expression and common elements currently used to predict prognosis (eg, T stage, N stage, or the American Joint Committee on Cancer stage grouping). Patients with tumors expressing higher levels of EGFR had significantly higher locoregional recurrences than those with lower EGFR-expressing tumors. Unexpectedly, EGFR expression was even a stronger predictor of locoregional control than T stage in multivariate analysis.
In this context, preclinical in vivo studies have demonstrated a close relationship between EGFR overexpression and cellular radioresistance in HNSCC.[18,19] Thus, EGFR blockade through a variety of strategies has the prospect of reducing radioresistance in patients with locally advanced HNSCC.
Targeting EGFR in HNSCC
Strategies for blocking EGFR function have targeted EGFR activation and/or phosphorylation primarily through the use of monoclonal antibodies to the receptor and tyrosine kinase-specific inhibitors. Discussed below are examples of strategies to block EGFR signaling:
Monoclonal Antibodies
EGFR-specific monoclonal antibodies have been developed with a higher affinity to the extracellular domain of the receptor than the natural ligand, acting as competitive inhibitors of EGFR. Cetuximab (C225, Erbitux), a human-mouse chimeric monoclonal antibody, has been widely studied preclinically. In addition to blocking EGF and TGF-alpha binding to EGFR, preventing receptor dimerization and autophosphorylation, cetuximab has been shown to affect proteins related to the induction of apoptosis or programmed cell death, including upregulation of p27 and BAX (proapoptotic protein) in vitro.[20,21]
Cetuximab also enhances the radiosenstivity of HNSCC.[22] This seems logical since we have learned that ionizing radiation induces a rapid activation of EGFR signaling. Through increased cell proliferation, promotion of DNA repair, and activation of the MAPK and PI3 kinase pathways, radioresistance is fostered.[ 23] The final result is a prosurvival response affected through an array of downstream transcription factors that enhance proliferation, angiogenesis, and metastasis.[23] Inhibition of EGFR signaling seems to reduce the ability of cells to recover after irradiation by arresting them in the G1 phase of the cell cycle.[24] The combination of cetuximab with radiation increases tumor apoptosis and decreases angiogenesis in cetuximab- responsive HNSCC tumor models.[22]
Tyrosine Kinase-Specific Inhibitors
Tyrosine kinase-specific inhibitors (TKIs) reversibly or irreversibly block EGFR autophosphorylation by competitively inhibiting the catalytic domain of EGFR. In vitro studies have demonstrated that gefitinib (ZD1839, Iressa) inhibits EGFR signaling through both Akt and MAPK pathways, causes G1 arrest, and suppresses growth of EGFR-expressing human cancer cell lines or xenografts. In regard to weight loss or death, gefitinib has synergistic effects when combined with chemotherapeutic agents and/or radiotherapy in mice, with minimal toxicity.[25,26] Many of these TKIs are also orally available, making them easy to administer and ideal for clinical trials. In addition to gefitinib, low- molecular weight TKIs in preclinical and clinical trials include CI-1033, an irreversible pan-erbB inhibitor, and erlotinib (OSI-774, Tarceva).[27]
Clinical Trials Targeting EGFR in HNSCC
Monotherapy
Many of the single-agent clinical trials have involved TKIs. A recent phase II trial of erlotinib enrolled 114 patients with advanced HNSCC refractory to chemotherapy.[27] Of the 78 patients evaluated for tumor response, 10 (23%) had a partial response, 23 (29%) had stable disease, and 45 (58%) had progressive disease. Observed toxicity included acne-like skin rushes (28% grade 1, 34% grade 2, 8% grade 3, and drug discontinuation in two patients), nausea, vomiting, headache, diarrhea, and fatigue.
A phase II trial of gefitinib monotherapy, focusing on recurrent or metastatic HNSCC, reported on 40 evaluable patients, 8 (20%) of whom experienced a complete or partial response. An additional 14 (35%) showed stable disease, and 18 (45%) experienced disease progression at 8 weeks.[28] Toxicity included grade 3 drug-related neurotoxicity (myelopathy), which resolved after termination of treatment. One patient discontinued therapy due to grade 2 acne-like rash, and three patients required dose reduction after experiencing mild-to-moderate diarrhea. Common side effects were grade 1/2 diarrhea (45%), acne-like rashes (45%), nausea (14%), and anorexia (20%). Of note, acne-like rash appeared to predict for response to gefitinib and was associated with improved survival. This association has not been clearly demonstrated in gefitinib monotherapy trials in other disease sites such as lung cancer.
CI-1033, an orally active, irreversible pan-erbB TKI, has undergone phase I testing to determine the doselimiting toxicities, minimal toxic dose, and pharmacokinetic profile.[29] Initial reports detailed the treatment of 34 advanced-stage cancer patients including 3 with head and neck cancer. Doselimiting hypersensitivity reactions occurred in two patients at 560 mg.
EMD 72000, a humanized monoclonal antibody against EGFR, has also been evaluated in patients with advanced malignancies, including patients with HNSCC. In this phase I study, 5 of 22 patients achieved a partial response, including 2 patients with heavily pretreated advanced HNSCC, resulting in an overall response rate of 23% (95% confidence interval: 8%-45%).[30] As in studies of cetuximab or gefitinib, no correlation between the degree of EGFR expression and tumor response to EMD 72000 was observed. Sufficient inhibition of the EGFR signaling pathway was achieved at EMD 72000 doses well below the minimal toxic dose.
Finally, ABX-EGF is a high-affinity, completely human IgG2 monoclonal antibody with strong activity against human EGFR. ABX-EGF prevents receptor binding of EGF/TGFalpha through competitive linking to the EGFR, inhibiting intracellular tyrosine phosphorylation and tumor cell activation. In A431 tumor models in nude mice, ABX-EGF prevented tumor formation and cured large, established tumors. Treatment with this agent alone, sans chemotherapy or radiotherapy, resulted in marked tumor growth inhibition at modest doses.[31]
EGFR Inhibition and Chemotherapy
In parallel with trials combining EGFR inhibitors and radiation for HNSCC, it is worth mentioning several studies that have combined EGFR inhibitors with platinum-based chemotherapy in advanced or recurrent HNSCC. Initial phase I trials established the loading dose of cetuximab at 400 mg/m2 with a maintenance dose at 250 mg/m2 as achieving a high percentage of EGFR saturation in tumor tissue.[32] Six (67%) of nine evaluable patients also achieved major responses, including two complete remissions.
The Eastern Cooperative Oncology Group (ECOG) performed a randomized phase III trial comparing cisplatin vs cisplatin and cetuximab.[ 33] A total of 121 patients with metastatic or recurrent HNSCC who had not received prior therapy for metastatic disease or induction or adjuvant therapy within 3 months were enrolled. Grade 3/4 hypersensitivity, neutropenia, and rash/desquamation occurred in 6%, 17%, and 11% of patients, respectively. Most patients had stable disease, with 15% of patients experiencing complete or partial response. At the last reported follow-up, the difference between the two arms with regard to survival was not statistically significant.
The use of cetuximab has also recently been investigated in combination with carboplatin in cisplatinrefractory patients with recurrent or metastatic nasopharyngeal cancer.[34] Ninety-three percent of tumors were EGFR-positive (56/60 patients). A partial response rate of 17% was reported, with an additional 42% of patients experiencing stable disease. A majority of the responses occurred within the first two cycles. A recent phase I trial in patients with recurrent or metastatic HNSCC combining cetuximab and cisplatin or carboplatin concurrently at fixed dose levels with escalating doses of fluorouracil (600, 800, and 1,000 mg/m2) reported 10 partial responses and 8 cases of stable disease.[35]
EGFR Antagonists and Radiotherapy
TABLE 1



Clinical Trials With EGFR-Signaling Inhibitors and Radiation Therapy in Patients With Head and Neck Cancer
Table 1 depicts some of the clinical trials that are ongoing or have been completed with EGFR inhibitors and radiation or chemoradiation in HNSCC. An early phase I study from the University of Alabama at Birmingham demonstrated that cetuximab could be given safely at a loading dose of 400 mg/m2 and a maintenance weekly dose of 250 mg/m2 with either conventional daily radiation (1.8 Gy per fraction) or 1.2 Gy per fraction, twice a day, to 76.8 Gy.[36] The majority of side effects were attributable to radiotherapy, and all patients in this study experienced an objective response.
A subsequent phase III study investigated the use of cetuximab combined with radiation vs radiation alone in locally advanced HNSCC. The toxicity and response results of this study were reported at the annual meeting of the American Society for Clinical Oncology (ASCO) in 2004. A statistically significant advantage was observed in favor of cetuximab plus radiation in terms of locoregional control at 2 years (P = .01), progressionfree survival, and overall survival. Of major importance was the fact that, overall, mucosal toxicity was not significantly higher than that seen in the radiotherapy-alone arm. These impressive initial findings without the use of conventional chemotherapeutics are provocative and perhaps suggest that the use of targeted agents and radiation should be tested against chemoradiation regimens in the future.[37]
At ASCO 2003, investigators from Memorial Sloan-Kettering Cancer Center reported on the toxicity and results of combined weekly cetuximab with concomitant boost radiation and cisplatin (100 mg/m2 IV, weeks 1 and 4) for patients with stage III/IV HNSCC.[38] Several reported grade 4 toxicities including anaphylaxis, myocardial infarction, and mucositis as well as two patient deaths were of concern. However, the results in the surviving patients were encouraging. Although this trial closed early due to toxicity, the 2-year actuarial and overall survival rates were both 76%, and 16 of 21 patients are disease-free at 26 months of median follow-up.[38]
Trials combining TKIs with radiation and cisplatin are currently under way, using both erlotinib and gefitinib in patients with advanced HNSCC. At the University of Colorado Comprehensive Cancer Center, in collaboration with the National Cancer Institute and several other participating institutions, an ongoing phase I trial is testing combinations of gefitinib concurrent with radiation in intermediate-stage disease, and gefitinib with radiation plus cisplatin (30 mg/m2/wk) in patients with stage IVA/ IVB disease.[39]
Alternative or Complementary EGFR Targeting Strategies
FIGURE 3

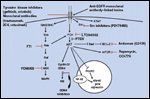
Options for Disrupting EGFR-Associated Signaling in Head and Neck Cancer
Many different approaches to disrupting upstream or downstream EGFR-associated activity are being studied (Figure 3). Along with the above mentioned strategies for EGFR inhibition, examples of additional attempts at EGFR signal perturbation warrant a brief discussion.
Anti-EGFR Antibody-Linked Toxins
Preclinical studies targeting EGFRoverexpressing cells using conjugates of toxins linked to anti-EGFR antibodies have shown some promise. The idea is that these "smart bombs" will selectively deliver the toxins to the receptor, either limiting cell proliferation by disrupting its signaling or killing the cell outright. A potential advantage of this approach is the possible "bystander effect" (in surrounding tumor cells) created with toxin delivery. Anti-HER2/neu Pseudomonas exotoxin has shown antitumoral efficacy when injected into HNSCC tumors in vivo.[40]
Alternatively, radioactive isotopes or chemotherapeutic agents can be used instead of toxins. Delivery of these anti-EGFR conjugates should be selective, as systemic therapy may result in EGFR-rich organs such as the liver becoming injured in the process. The opportunity to combine these molecules with radiation is intriguing.
STAT3 Targeting
STATs are transcription factors involved in the regulation of gene expression and associated with EGFR signaling. Seven STAT proteins have been identified, and STATs 1, 3, and 5 are constitutively activated in HNSCC overexpressing EGFR.[41] In HNSCC, constitutive activation of STAT3 and STAT5 has been linked to TGF-alpha/EGFR signaling in vitro and in vivo.[41] Reduction of STAT3 has been shown to inhibit the growth of HNSCC.[42] Although frequently associated with EGFR activation, STAT3 may also act independently of EGFR signaling in head and neck cancer and is amplified in some HNSCC lines that have low EGFR expression.[43] This appears to be related to its interaction with interleukin-6 cytokines in a separate autocrine/paracrine loop. Perhaps downstream inhibition of STAT3 might complement upstream blockade of EGFR activation and improve response to EGFR targeting.
mTOR Inhibition
Targeting downstream proteins associated with EGFR activation may improve response to EGFR inhibitors. In this regard, the mammalian target of rapamycin (mTOR) belongs to a recently identified family of protein kinases termed phosphoinositide 3- kinase related kinases (PIKKs) downstream of the EGFR, which are involved in cell-cycle progression and checkpoints that govern cellular responses to DNA damage, repair, and recombination.[44]
Compounds designed to interfere with this molecule include CCI-779, a clinically relevant analog of rapamycin. Both agents block cell-cycle progression from G1 to S phase through interference with mRNA translation of cell-cycle proteins. Inactivating cyclin/cyclin-dependent kinase/cyclin- dependent kinase inhibitors by blocking cancer cells in the early phases of the cell cycle enables mTOR inhibitors to stop the growth of cancer cells.[45]
Hurdles to Overcome
Predicting Response to EGFR Inhibitors
If EGFR inhibitors are to be relevant in the treatment of HNSCC, we need to determine exactly how they work. A key issue is whether EGFR expression is a relevant predictor of response; a cancer may not necessarily rely on EGFR signaling alone for its survival. In addition, the integrity of the EGFR-activated downstream intracellular signal transduction machinery may influence the response to these drugs. Recent experimental evidence suggests that cancer cells may escape growth inhibition by using alternative growth pathways or by constitutive activation of downstream signaling effectors.
Despite the promising preclinical findings seen with EGFR blockade in many epithelial malignancies, the clinical response in pretreated HNSCC patients receiving EGFR inhibitors has hovered at ~15%. Why haven't we observed a higher response when so many HNSCC cancers express EGFR? The current thought is that a host of molecular abnormalities play a role in determining head and neck carcinogenesis and biologic behavior. Therefore, similar histologic types notwithstanding, the activated cell survival pathways may vary among HNSCCs. Differences in activated EGFR-associated phosphoinositide-3-kinase (PI3K), MAPK, or STAT3 may account for the differences in tumor response to EGFR antagonists. This hypothesis can be partially addressed by a panel of correlative biomarker studies examining the association between the expression profiles of these molecules and the tumor response, along with other as of yet unknown proteins/genes, using tumor specimens of patients enrolled into prospective trials.
Understanding which HNSCC cancers will respond to EGFR inhibition may also relate to the expression and activity of the remaining family members of EGFR (erbB2-4). Also, the presence of exogenous EGF-related ligands may, in part, govern tumor response to anti-EGFR agents, and thus, dual or multiple blocking of other family members of EGFR may be required to achieve the desired effects.[ 46] Increased expression levels or mutations in downstream proteins such as Ras, Raf, PTEN, and Akt may also predict response, along with other as yet unknown proteins/genes, independent of EGFR expression, as shown in Figure 4.
FIGURE 4


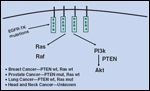
Mutated Elements of the EGFR-Kinase Pathway in Tumors May Predict for Response to EGFR Inhibitors
An emerging issue is whether rash is predictive of response to EGFR inhibitors as well as survival. Across the board in phase II studies employing cetuximab-including a trial in patients with advanced HNSCC-patients who developed the acne-like rash survived longer than those who did not develop a rash, and the more intense the rash, the longer the survival. The findings from four phase II studies incorporating cetuximab, as presented by Saltz et al at ASCO 2003, suggests that skin rash may be an important surrogate predictor of efficacy.[ 47] Similar findings were seen in a phase II study in advanced HNSCC patients treated with gefitinib monotherapy.[ 28] This observation has not held up in stage IV lung cancer patients treated with gefitinib. Perhaps individualizing patient dosing to stimulate skin rash will be more closely evaluated in future clinical trials with EGFR inhibitors.
Preventing Resistance to EGFR Inhibition
Recent experimental evidence suggests that cancer cells may escape growth inhibition by using alternative growth or angiogenic pathways. For example, resistance to anti-EGFR antibodies such as cetuximab and hR3 has been reported in A431 tumors in mice. Overexpression of vascular endothelial growth factor (VEGF) may have been a contributor to resistance through the upregulation of angiogenesis.[48] Activation of the insulin-like growth factor receptor I contributed to continuous activation of the antiapoptotic PI3K-signaling pathway that blocked EGFR inhibitors such as gefitinib in human glioblastoma cells in vitro.[49]
These observations, along with increasing evidence that superfluous growth pathways are active in neoplastic cells, form the basis for testing therapeutic strategies targeting multiple pathways. Indeed, preclinical studies have shown significant and sustained antitumor activity in vitro and in vivo by combining anti-EGFR agents with, for example, inhibitors of the cAMP-dependent protein kinase (type I PKA)[50] or a VEGF antisense oligonucleotide.[51] Other promising strategies have included concurrent blockage of the VEGF receptor (VEGFR) using PTK 787 and EGFR by PKI 166, both tyrosine kinase inhibitors directed at VEGFR and EGFR signaling respectively in a pancreatic model,[52] and the combination of gefitinib with the anti-erbB2 antibody trastuzumab (Herceptin).[53] Figure 3 presents possible polytargeted scenarios against HNSCC.
An alternative method that might prevent the development of resistance to EGFR blockade would be to administer single agents with dual inhibitory action toward EGFR and VEGFR signaling. ZD6474 represents such an agent. It is a small-molecule inhibitor of KDR/VEGFR-2 tyrosine kinase activity and of a variety of other tyrosine and serine-threonine kinases including those activated by EGFR.[54] In animals bearing human colon cancer xenografts with acquired resistance to gefitinib or cetuximab, ZD6474 administration resulted in significant tumor growth inhibition.[55] Tumor cells resistant to cetuximab or gefitinib exhibited a marked increase in activated MAPK, cyclooxygenase- 2, and VEGF compared with the control GEO cells. These data indicate that inhibition of VEGF signaling has potential as an anticancer strategy, even in tumors that are resistant to EGF inhibitors.
Another attractive approach applicable to the treatment of HNSCC might include the combination of two molecules acting on different domains of EGFR. Gefitinib plus cetuximab provided supra-additive cancer growth inhibition on the high EGFR-expressing A431 tumor in vitro and in vivo (P < .05) [56]. In this particular model, 25 mg/kg of gefitinib plus cetuximab resulted in a 90% growth inhibition and induced complete remissions in 3 out of 10 tumors, and 50 mg/kg of gefitinib plus cetuximab resulted in 100% complete remission. This suggests that hindering both the extracellular and intracellular activation sites of EGFR may prevent the cancer cell from overriding a specific blockade.
Because other EGFR family members may play a role in HNSCC, preventing coupling of, for example, erbB2 to EGFR (or erbB1) might prevent redundant signaling from overriding or bypassing EGFR blockade. In this regard, 2C4-which binds to a different portion of the extracellular domain than trastuzumab (Herceptin) and prevents the receptor from dimerizing with other erbB family members- is under investigation and appears active irrespective of EGFR status.[57,58] Further studies assessing the role of 2C4 in polytargeted therapy against HNSCC are ongoing.
Sequencing Issues With EGFR Inhibitors
One of the concerns recently raised is the potential antagonistic effects that might be seen with concurrent administration of TKIs and chemotherapy. Several clinical trials combining TKIs with chemotherapy in the treatment of advanced lung cancer failed to show a survival benefit over chemotherapy alone.[59,60] It is unclear whether there are differences in interactions with antibodies vs TKIs and chemotherapy and whether the type of chemotherapy dictates synergy or antagonism.
An early report by the Lung Cancer CetuximAb Study (LUCAS), presented at ASCO 2003,[61] evaluated the response rate of the combination of cetuximab plus cisplatin/vinorelbine or of the same chemotherapy alone as first-line treatment in patients with EGFR-positive stage IIIB/IV NSCLC. The overall response rates were 50% (nine partial responses, eight confirmed; seven cases of stable disease; and two cases of progressive disease) in arm A and 29% (five partial responses, three confirmed; six cases of stable disease; and six cases of progressive disease) in arm B. The trial continues to accrue patients with the intent of recruiting a total of 40 patients per arm. It will be interesting to see if these response rates hold up with mature follow-up. Limited information is available on sequencing issues using EGFR inhibitors and radiation, and this is an area that warrants further preclinical and clinical investigation.
Conclusions
In this review, we have tried to provide a review of the emerging role of targeted therapies in HNSCC. The goal of targeted therapy should include the development and testing of agents with selective activity against HNSCC and low systemic toxicity. Ideally, a molecular target in HNSCC meets the following criteria: (1) it drives tumor growth, (2) it demonstrates reversible function by pharmacologic inhibition, (3) its inhibition is tolerated by normal cells, and (4) effects on it are measurable in tumor tissue.
We are clearly establishing new ground in the fight against HNSCC with the introduction of drugs that attack specific parts of the cancer cell growth pathways. Despite the gains achieved with concurrent chemoradiotherapy, the toxicities can be daunting at times. Thus, the foundations established with EGFR inhibitors and radiotherapy should embolden oncology investigators to design clinical trials that incorporate combinations of targeted agents with different mechanisms of action. An additional task is to return to the laboratory and understand why EGFR inhibitors work, why they are active in EGFR-negative as well as EGFR-positive tumors, how to better predict response, and the optimal way to sequence these drugs with radiation. Advancement in these areas will lead to optimal stratification or selection of patients based on tumor or serum markers, for testing promising novel multitargeted therapy regimens in clinical trials.
Financial Disclosure:Dr. Raben has received partial financial support for preclinical studies conducted through the University of Colorado Comprehensive Cancer Center from AstraZeneca.
References:
1. Sessions R, Harrison L, Forastiere A: Tumors of the larynx and hypopharynx, in DeVita Jr VT, Hellman S, Rosenberg SA (eds): Cancer: Principles and Practice of Oncology. Philadelphia, Lippincott-Raven, 1997.
2. Parkin D, Pisani P, Ferlay J: Global cancer statistics. CA Cancer J Clin 49:33-64, 1999.
3. Vokes E, Weichselbaum R, Lippman S, et al: Head and Neck Cancer. N Engl J Med 328:184-194, 1993.
4. Cancer: Adjuvant chemotherapy for advanced head and neck squamous carcinoma. Final Report of the Head and Neck Contracts Program. Cancer 60:301-311, 1987.
5. Santini J, Formento J, Francoual M, et al: Characterization, quantification, and potential clinical value of the epidermal growth factor receptor in head and neck squamous cell carcinomas. Head Neck 13:132-139, 1991.
6. Ang K, Berkey B, Tu X, et al: Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 62:7350-7356, 2002.
7. Modjtahedi H, Dean C: The receptor for EGF and its ligands: Expression, prognostic value and target for therapy in cancer (review). Int J Oncol 4:277-296, 1994.
8. Thomas S, Grandis J: Pharacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat Rev 30:255-256, 2004.
9. Ibrahim S, Vasstrand E, Liavaag P, et al: Expression of c-erbB protonocogene family members in squamous cell carcinoma of the head and neck. Anticancer Res 17: 4539-4546, 1997.
10. Xia W, Lau Y, Zhang H, et al: Strong correlation between c-erbB-2 overexpression and overall survival of patients with oral squamous cell carcinoma. Clin Cancer Res 3:3-9, 1997.
11. Shintani S, Funayama T, Yoshihama Y, et al: Prognostic significance of ERBB3 overexpression in oral squamous cell carcinoma. Cancer Lett 95:79-83, 1995.
12. Grandis J, Tweardy D: Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res 53:3579-3584, 1993.
13. Rubin Grandis J, Melhem M, Barnes E, et al: Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer 78:1284-1292, 1996.
14. Rubin Grandis J, Zeng Q, Tweardy D: Retinoic acid normalizes the increased gene transcription rate of TGF-alpha and EGFR in head and neck cancer cell lines. Nature Med 2:237-240, 1996.
15. Hendler F, Shum-Siu A, Nanu L, et al: Overexpression of EGF receptors in squamous tumors is associated with poor survival. J Cell Biochem 12:105, 1988.
16. Miyaguchi M, Olofosson J, Hellquist H: Expression of epidermal growth factor receptor in glottic carcinoma and its relation to recurrence after radiotherapy. Clin Otolaryngol 16:466-469, 1991.
17. Rubin Grandis J, Melhem MF, Gooding WE, et al: Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst 90:824- 832, 1998.
18. Akimoto T, Hunter N, Buchmiller L, et al: Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res 5:437-443, 1999.
19. Balaban N, Moni J, Shannon M, et al: The effect of ionizing radiation on signal transduction: Antibodies to EGF receptor sensitize A431 cells to radiation. Biochimica et Biophysica Acta 1314:147-156, 1996.
20. Mendelsohn J: Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy. Clin Cancer Res 3:2703- 2707, 1997.
21. Huang S, Bock J, Harari P: Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res 59:1935-1940, 1999.
22. Huang S, Harari P: Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res 6:2166-2174, 2000.
23. Dent P, Reardon D, Park J, et al: Radiation- induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation- induced cell death. Mol Biol Cell 10:2493-506, 1999.
24. Harari P, Huang S: Head and neck cancer as a clinical model for molecular targeting of therapy: Combining EGFR blockade with radiation. Int J Radiat Oncol Biol Phys 49:427- 433, 2001.
25. Ciardiello F, Caputo R, Bianco R, et al: Antitumor effects and potentiation of cytotoxic drugs: Activity in human cancer cells by ZD1839 (Iressa), and epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 6:2053-2063, 2000.
26. Sirotnak F, Zakowski M, Miller V, et al: Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res 6:4885-4892, 2000.
27. Senzer N, Soulieres D, Siu L, et al: Phase 2 evaluation of OSI-774, a potent oral antagonist of the EGFR-TK in patients with advanced squamous cell carcinoma of the head and neck (abstract 6). Proc Am Soc Clin Oncol 20:2a, 2001.
28. Cohen E, Rose F, Stadler W, et al: Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol 21:1980-1987, 2003.
29. Shin D, Nemunaitis J, Zinner R, et al: A phase I clinical and biomarker study of CI- 1033, a novel pan-ErbB tyrosine kinase inhibitor in patients with solid tumors (abstract 324). Proc Am Soc Clin Oncol 20:82a, 2001.
30. Vanhoefer U, Tewes M, Rojo F, et al: Phase I study of the humanized antiepidermal growth factor receptor monoclonal antibody EMD72000 in patients with advanced solid tumors that express the epidermal growth factor. J Clin Oncol 1:175-184, 2004.
31. Foon K, Yang X, Weiner L, et al: Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys 58:984-990, 2004.
32. Shin D, Donato N, Perez-Soler R, et al: Epidermal growth factor receptor-targeted therapy with C225 and cisplatin in patients with head and neck cancer. Clin Cancer Res 7:1204- 1213, 2001.
33. Burtness B, Li Y, Flood W, et al: Phase III trial comparing cisplatin (C) + placebo (P) to C + anti-epidermal growth factor antibody (EGF-R) C225 in patients (pts) with metastatic/ recurrent head & neck cancer (HNC) (abstract 901). Proc Am Soc Clin Oncol 21:226a, 2002.
34. Chan A, Hsu M, Goh E, et. al.: A phase II study of cetuximab (C225) in combination with carboplatin in patients (pts) with recurrent or metastatic nasopharyngeal carcinoma (NPC) who failed platinum based chemotherapy (abstract 2000). Proc Am Soc Clin Oncology 22:497, 2003.
35. Vega-Villegas E, Awada A, Mesia R, et al: A phase I study of cetuximab in combination with cisplatin or carboplatin and 5-fluorouracil (5-FU) in patients (pts) with recurrent or metastatic squamous cell carcinoma of the head and neck (abstract 2020). Proc Am Soc Clin Oncol 22:502, 2003.
36. Robert F, Ezekiel M, Spencer S, et al: Phase I study of anti-epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol 19:3234-3243, 2001.
37. Bonner JA, Biralt J, Harari PM, et al: Cetuximab prolongs survival in patients with locoregionally advanced squamous cell carcinoma of head and neck: A phase III study of high dose radiation therapy with or without cetuximab (late-breaking abstract 5507). Proceedings of the American Society of Clinical Oncology 40th Annual Meeting, New Orleans, 2004.
38. Pfister D, Aliff T, Kraus D, et al: Concurrent cetuximab, cisplatin and concomitant boost radiation therapy (RT) for locoregionally advanced, squamous cell head and neck cancer (SCHNN): Preliminary evaluation of a new combined modality paradigm (abstract 1993). Proc Am Soc Clin Oncol 22:495, 2003.
39. Raben D, Weng E, Kane M, et al: Preliminary report on toxicity of a phase I trial of gefitnib (Iressa) in combination with radiation/ chemotherapy for patients with locally advanced head and neck cancer (LAHNC) (abstract C186). Proceedings of the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics, Boston, 2003.
40. Azemar M, Schmidt M, Arlt F, et al: Recombinant antibody toxins specific for ErbB2 and EGF receptor inhibitors in vitro growth of human head and neck cancer cells and cause rapid tumor regression in vivo. Int J Cancer 86: 269-275, 2000.
41. Grandis J, Drenning S, Chakraborty A, et al: Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest 102:1385-1392, 1998.
42. Song J, Grandis J: STAT signaling in head and neck cancer. Oncogene 19:2489- 2495, 2000.
43. Sriuranpong V, Park J, Amornphimoltham P, et al: Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin6/gp130 cytokine system. Cancer Res 63:2948-2956, 2003.
44. Sarkaria J, Tibbetts R, Busby E, et al: Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 58:4375-4382, 1998.
45. Hidalgo M, Rowinsky E: The rapamycinsensitive signal transduction pathway as a target for cancer therapy. Oncogene 19:6680-6686, 2000.
46. Motoyama A, Hynes N, Lane H: The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res 62:3151-3158, 2002.
47. Saltz L, Kies M, Abbruzzese J, et al: The presence and intensity of the cetuximab-induced acne-like rash predicts increased survival in studies across multiple malignancies (abstract 817). Proc Am Soc Clin Oncol 22:204, 2003.
48. Viloria-Petit A, Crombet T, Jothy S, et al: Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo. Cancer Res 61:5090-5101, 2001.
49. Chakravarti A, Loeffler J, Dyson N: Insulin- like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res 62:200-207, 2002.
50. Ciardiello F, Caputo R, Bianco R, et al: Cooperative inhibition of renal cancer growth by anti-epidermal growth factor receptor antibody and protein kinase A antisense oligonucleotide. J Natl Cancer Inst 90:1087-1094, 1998.
51. Ciardiello F, Bianco R, Damiano D, et al: Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor. Clin Cancer Res 6:3739-3747, 2000.
52. Baker C, CC S, Fidler I: Blockade of vascular endothelial growth factor receptor and epidermal growth factor receptor signaling for therapy of metastatic human pancreatic cancer. Cancer Res 62:1996-2003, 2002.
53. Normanno N, Campiglio M, De Luca A, et al: Cooperative inhibitory effect of ZD1839 (Iressa) in combination with trastuzumab (Herceptin) on human breast cancer cell growth. Ann Oncol 13:65-72, 2002.
54. Wedge S, Ogilvie D, Dukes M, et al: ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 62:4645-4655, 2002.
55. Ciardiello F, Bianco R, Caputo R, et al: Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res 10:784-793, 2004.
56. Matar P, Rojo R, Guzman M, et al: Combined anti-epidermal growth factor receptor (EGFR) treatment with a tyrosine kinase inhibitor gefitinib (ZD 1839, "Iressa") and a monoclonal antibody (IMC-C225): Evidence of synergy. Proc AACR 44:917, 2003.
57. Agus D, Akita R, Fox W, et al: Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2:127-137, 2002.
58. Baselga J: A new anti-ErbB2 strategy in the treatment of cancer: Prevention of liganddependent ErbB2 receptor heterodimerization. Cancer Cell 2:93-95, 2002.
59. Giaccone G, Johnson D, Manegold C, et al: A phase III clinical trial of ZD1839 (Iressa) in combination with gemcitabine and cisplatin in chemotherapy-naive patients with advanced non-small-cell lung cancer (INTACT 1). Ann Oncol 13:2, 2002.
60. Johnson D, Herbst R, Giaccone G, et al: ZD1839 ("Iressa") in combination with paclitaxel and carboplatin in chemotherapy-naive patients with advanced non-small-cell lung cancer (NSCLC): Results from a phase III clinical trial (INTACT 2). Ann Oncol 13:127, 2002.
61. Gatzemeier U, Rosell R, Ramlau R, et al: Cetuximab (C225) in combination with cisplatin/vinorelbine vs. cisplatin/vinorelbine alone in the first-line treatment of patients (pts) with epidermal growth factor receptor (EGFR) positive advanced non-small-cell lung cancer (NSCLC) (abstract 2582). Proc Am Soc Clin Oncol 22:642, 2003.












