Emerging Role of Immunotherapy in the Management of Prostate Cancer
There has been a resurgence of interest in developing noncytotoxic immune therapies for patients with either hormone-naive biochemically relapsed post-primary therapy or castrate metastatic prostate cancer. The rationale for developing an immunotherapeutic approach has been based on the overexpression and underglycosylation of a wide variety of altered "self" molecules including prostate-specific antigen (PSA), acid phosphatase (ACP), prostate stem cell antigen (PSCA), and prostate-specific membrane antigen (PSMA), which can serve as targets for immune recognition and attack. In addition, such a strategy could theoretically make use of the patient's immune system to fight the tumor particularly if their disease is of reasonably low volume. A variety of immunotherapeutic approaches have been explored through phase I, II, and now phase III trials demonstrating that immunologic tolerance could be broken, as evidenced by the development of high-titer antibodies and T-cell responses specific for the tumor. What appears to be revolutionizing the immunotherapy field is the combination of vaccines with cytokines or immune modulators, which not only potentiate immune reactivity in vivo but foster dramatic antitumor responses. This review explores the challenges now faced in establishing a role for immune therapies for prostate cancer treatment.
There has been a resurgence of interest in developing noncytotoxic immune therapies for patients with either hormone-naive biochemically relapsed post-primary therapy or castrate metastatic prostate cancer. The rationale for developing an immunotherapeutic approach has been based on the overexpression and underglycosylation of a wide variety of altered "self" molecules including prostate-specific antigen (PSA), acid phosphatase (ACP), prostate stem cell antigen (PSCA), and prostate-specific membrane antigen (PSMA), which can serve as targets for immune recognition and attack. In addition, such a strategy could theoretically make use of the patient's immune system to fight the tumor particularly if their disease is of reasonably low volume. A variety of immunotherapeutic approaches have been explored through phase I, II, and now phase III trials demonstrating that immunologic tolerance could be broken, as evidenced by the development of high-titer antibodies and T-cell responses specific for the tumor. What appears to be revolutionizing the immunotherapy field is the combination of vaccines with cytokines or immune modulators, which not only potentiate immune reactivity in vivo but foster dramatic antitumor responses. This review explores the challenges now faced in establishing a role for immune therapies for prostate cancer treatment.
Therapeutic antitumor immunity or immunotherapy for prostate cancer has been an attractive treatment option for all stages of prostate cancer, given its seemingly broad application and low toxicity profile. Its appeal has been based on the idea that the evolution of cancer is due to a defect in immune surveillance and that the body can use other aspects of its immune system to fight the cancer. Vaccination against cancer is not new-in fact, the front page of The Globe from Toronto on July 17, 1925, boasted of a novel report by Gye and Barnard in the Lancet suggesting that a virus caused cancer and that "while it would be an exaggeration to say a cure for cancer is in sight, the new announcement is expected to indicate probabilities of finding a method of vaccination against cancer."[1]
Have we made strides since that time? A variety of precedents have established the success of vaccines in certain cancers, especially melanoma. We now know that some anti-inflammatory responses and autoimmune phenomena, as seen with depigmentation or vitiligo, can herald antitumor immunity. Many who have followed the immunotherapy literature would say the development of such approaches has run the gamut of investigation, from whole-cell vaccines to cell extracts, to purified membranes expressing the antigen, to purified antigen, to protein, peptides, and ultimately DNA.
Given the diversity of expression of multiple antigens on a cancer cell surface, it remains unclear which of these are specifically associated with the cancer and should be the appropriate target(s) for therapeutic direction. Despite multiple approaches, no one approach has been shown to have a superior impact on slowing cancer growth or inducing disease remission. Antigens expressed on tumor cells may be "self" antigens-molecules derived from the breakdown of normal cell membranes or remnants of cells infected by or destroyed by bacteria or viruses or tumor cells. Those abnormal antigens are taken up by scavenger cells known as antigen-presenting cells or dendritic cells (called Langerhans cells in the skin), digested, and broken down into peptides, which are then presented to the T cell for recognition and, hopefully, subsequent destruction.
Much of what we have learned in designing immunotherapeutic approaches has been based on antigens isolated from infectious diseases. An antigen, while foreign to the immune system, may be insufficient to generate an immune response. Not only must it be immunogenic, but the immune system must "see" it in order to determine whether recognition is feasible. In the cases of altered self antigens, the immune system may see this molecule as self and therefore not be triggered to induce a response. However, if one were to try to make the immune system become aware of this altered molecule, immunizing with this isolated molecule may be insufficient and mandate that something be done to make the body react to it. This includes using a carrier molecule to make the molecule look larger in addition to revving up the immune system with a natural or synthetic chemical known as an adjuvant.
Induction of Immunity
Active vs Passive Approaches
Immunity to a particular molecule can be generated either passively or actively. In the former case, the immune system is not actively responding to the immunogen administered. In lieu of the body's own reaction, a drug such as a monoclonal antibody-which exquisitely targets a particular antigen-has already been generated, and one is "passively" conferred the immunity against that molecule by the antibody. Active immunity is the body's own response to a treatment, that is, the ability to generate a humoral or cellular immune response to an immunogen. Each subserves a different purpose in generating a reaction to tumor antigens.
Passive Immunity: Immunotherapy With Monoclonal Antibodies
Prostate-specific membrane antigen (PSMA) is one of several self antigens that has served as a potential target for immunologic approaches, with strategies including DNA and protein vaccines in addition to monoclonal antibodies.[2-11] PSMA is a type II integral transmembrane protein and a member of a superfamily of zinc-dependent exopeptidases, including carboxypeptidases A and G2, and peptidases T and V.[2-4] While highly expressed in prostate cancer cells and initially thought to be a specific marker for prostate cancer, its expression has been found in the brain and salivary gland.[2] It is also expressed in nonprostatic solid tumor neovasculature (eg, renal cell carcinoma),[5-7] making it a reasonable target for immune therapies such as vaccine strategies[8] and radiopharmaceutical targeting with monoclonal antibodies.[9-14]
As a type II transmembrane glycoprotein, PSMA is a large extracellular globular-like molecule with an intracellular segment. It is expressed at levels over a 1,000-fold greater than the minimal expression seen in the kidney, proximal small intestine, and salivary gland. It is also upregulated in the androgen-resistant state. Second-generation humanized forms of a monoclonal antibody against the external domain of PSMA (J591) developed by Bander et al[12,14] have been conjugated to iodine and yttrium, as well as to lutetium in an effort to image all sites of metastases, especially in bone. These studies have shown 100% specificity and sensitivity for sites in bone and have been extended to other malignancies including renal cell cancer.
• Clinical Trials-Two recent trials show good tolerability of alternative radiopharmaceuticals that have been coupled to J591 monoclonal antibody. Bander et al[14] performed a phase I study in 35 patients with androgen-independent prostate cancer to assess the safety, maximum tolerated dose (MTD), pharmacokinetics, organ dosimetry, targeting, human anti-J591 response, and biologic activity of lutetium-177-labeled J591. Of the 35 patients, 16 received up to three doses. Myelosuppression was dose-limiting at 75 mCi/m2, with the MTD established at 70 mCi/m2. Targeting to all known sites of disease (ie, bone and lymph nodes) was seen in all 30 patients. No patient developed a human anti-J591 antibody response to dehumanized J591 irrespective of the number of doses. Biologic activity lasting 3 to 8 months was seen in four patients, who had a at least a 50% decline in prostate-specific antigen (PSA) levels. Biomarker stabilization was seen in 16 patients for a median of 60 days.
Morris et al[13] also studied unlabeled and indium-111 (111In)-labeled J591 in a similar population. Two groups of seven patients with androgen-independent disease were studied. One group of patients received unlabeled J591 before the labeled antibody; the other received both together. The agent was well-tolerated, and biodistribution of 111In-labeled J591 was comparable in both groups, with half-lives of 0.96, 1.9, 2.75, and 3.47 days for the 10-, 25-, 50-, and 100-mg doses, respectively. Hepatic saturation occurred by the 25-mg dose. Antibody-dependent cell-mediated cytotoxicity was proportional to dose, with one patient showing a greater than 50% decline in PSA.
Tumor Antigens
Self antigens identified on prostate cancer cells and cell lines appear on the normal counterparts of the tumor cells derived from the organ in question, yet change in very subtle ways with malignant transformation.[15] This is particularly true of the mucin family (ie, MUC-1 and MUC-2, which are present normally within glandular elements and become overexpressed and underglycosylated with malignancy). It is unclear why the immune system does not see these altered self antigens expressed on the new tumor cells as foreign and enables them to grow. However, strategies have focused on means of educating the immune system to recognize these molecules and break immunologic tolerance, thereby allowing the body to respond to what is then perceived as a foreign rather than a self antigen.
Active Immunity: Augmenting Immunogenicity by Enhancing the Humoral Immune System
Vaccines designed to induce an optimal antibody response have several components, each of which must be optimized (Table 1).[15-19] The first component is the antigen itself, which must closely resemble its expression on the target-for example, Thomsen-Friedenreich (TF) antigen expression on tumor mucins. The TF antigen is a disaccharide covalently attached to serine or threonine (akin to the closely associated disaccharide sTn). Monoclonal antibodies and sera selected for preferential reactivity with cancer cells (as opposed to normal tissues) react with clusters of three such disaccharide antigens rather than a single antigen.[19]



The second prerequisite for an optimal antibody response is that the antigen is covalently conjugated to an immunogenic carrier protein. We have found keyhole limpet hemocyanin (KLH) to be the optimal carrier for antibody induction.[15-19] The conjugation must be achieved in a manner that does not interfere with the antigenic epitope itself (TFc) and that achieves as high an antigen/carrier ratio as possible (in this case, 466 or more TFc molecules per KLH molecule).[19] This was achieved using a hetero-bifunctional cross-linker, which links the terminal cysteine group of the cluster backbone to amino groups on KLH.
The final necessary component is the immunologic adjuvant. In our experience, saponin adjuvants such as QS21 have been the most potent for augmenting the antibody response against conjugate vaccines. The 100-µg dose level of QS21 used here has been found to be optimal, with higher doses resulting in excessive local and systemic toxicity and lower doses resulting in decreased immunogenicity.[15-19]
How to Measure a Biologic Response: Does an Immune Response Correlate With an Antitumor Effect?
Many different strategies have been undertaken toward developing an "ideal" immune-mediated therapy for treating prostate cancer. Many of these approaches have used prostate cancer cell lines such as LnCAP or PC-3, which have been genetically transduced with cytokine genes such as interleukin(IL)-2 or granulocyte-macrophage colony-stimulating factor (GM-CSF) in order to enlist ancillary immune cell recruitment. Alternative approaches include the transfection of prostate cancer cell lines infected with viral vectors such as fowlpox,[20] vaccinia,[21] adenoviruses,[22] or plasmids,[23] which enhance antibody and potentially T-cell responses. Others have demonstrated that synthetic mimes of known altered self antigens overexpressed on the prostate cancer cell surface can elicit specific immune responses when coupled to carriers such as KLH and given with QS21, but were unable to induce T-cell immunity.[16-19]
One thing in common with all these approaches is that while vigorous antibody responses can be generated, little or no antitumor response has been seen in patients with high tumor volumes. Moreover, no criteria have been established to better define what should be considered a response to the cancer. That is, are there specific biologic/immunologic parameters that correspond with disease response, or are newer criteria needed to assess what "response" really means? Another limitation to these approaches is that there is no easy way to potentiate and quantitate a T-cell immunity, which most immunologists feel is critical to enhancing and assessing antitumor responsiveness, respectively.
Finally, it remains unclear which antigen(s) is/are the "right target(s)" and which patient population would benefit from these approaches. It has been thought that the heavier the tumor burden, the greater the likelihood that the immune system will be suppressed and that patients with minimal disease would benefit. However, there is no definitive way to quantitate a clinical response to vaccine therapy in patients who have biochemically relapsed following definitive primary therapy such as surgery or radiation.
Any decline in PSA may not reflect a change in the biology of the tumor, and in vitro evidence of antitumor immunity may not be a realistic reflection of what is taking place in vivo. The use of PSA log slopes as a measure of antitumor effect is under consideration as a means of assessing antitumor effect, but this approach remains to be validated.[16,24] Several other studies have demonstrated similar effects with PSA slopes using vaccines comprised of dendritic cells [25] or genetically altered prostate cancer cell lines given with systemic cytokines such as GM-CSF.[26-30] The clinical impact of stabilization or decline of posttreatment vs pretreatment PSA log slopes and its relevance as an intermediate endpoint remains to be validated.
The Cellular Immune System: How to Better Harness Effector Cells
Multiple vaccine approaches have suggested that while immunologic tolerance could be broken, individual vaccines could not sufficiently effect an antitumor response, as evidenced by continued disease progression. Therefore, the combination of a vaccine with a biologic modulator, cytokine, or checkpoint inhibitor might be a reasonable option. Vaccines derived from whole-cell, purified antigen, peptide, or DNA have demonstrated that immunologic tolerance could be successfully broken, as evidenced by the generation of high-titer antibodies with specificity for the immunogen but often weak or no T-cell responses. However, these responses do not necessarily correlate with antitumor effects such as disease regression or decline in biomarkers such as PSA.
Immunology aficionados felt that enhancement of T-cell responses would be more specific and that recruitment of specific T-cell subpopulations was necessary to induce a more active and durable response. Attempts to integrate this approach into the clinic have already begun based on the success of preclinical trials.
One new approach toward enhancing T-cell responsiveness either alone or in conjunction with a vaccine has been through blocking or elaborating costimulatory molecules, which have been used in T-cell recognition, proliferation, and killing. Although attempts have been made to manipulate T cells as a major component of immune responses to a wide range of immunologic therapies, less than optimal results may have been due to technical difficulties in assessing T-cell responses in the laboratory as well as limitations in understanding the normal regulatory processes that limit T-cell responses to avoid autoimmunity. While T-cell responses are initiated by T-cell antigen receptor signaling in the context of an antigen within the pocket of major histocompatibility complex (MHC) on the cell surface, multiple other cell surface molecules also participate in a complex interplay with cytokines, which can act in a stimulatory or inhibitory manner.



Our understanding of the complexity of these regulatory pathways was increased with the demonstration by several groups that B7.1 and B7.2 (Table 2, Figure 1), whose expression is limited to "professional" antigen-presenting cells (APCs), can also interact with another molecule cytotoxic T-lymphocyte-associated protein 4, or CTLA-4 (Figure 1).[31] Allison's group[32-37] identified this protein on the T-cell surface, where it suppresses the ability of T cells to attack cancer cells. While initially thought to be another costimulatory molecule, CTLA-4 was shown to be an inhibitory molecule that functioned as a "checkpoint" limiting T-cell activation and expansion. It also has been shown to play a critical role in preventing or enhancing autoimmunity in several animal systems.


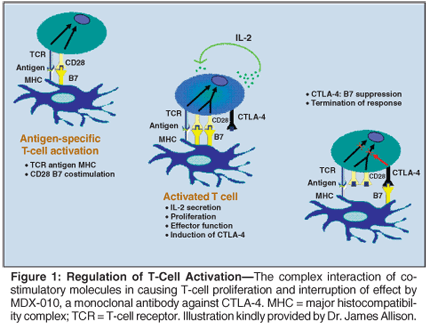
Prior studies using multiple experimental tumors in mice showed that CTLA-4 blockade could enhance antitumor responses either as a single agent or in combination with a vaccine.[38-40] CTLA-4 blockade acted synergistically with conventional cytotoxic therapies including vaccines in patients with prostate cancer, melanoma, renal cell carcinoma, or ovarian cancer. One study demonstrated significant synergism when anti-CTLA-4 antibody was given with GVAX to a patient with recurrent ovarian cancer.[41] Rechallenge of the patient with CTLA-4 after receiving prior vaccine resulted in decline in CA-125 for over 2 years, with stabilization of disease.
Although there have been instances of autoimmune breakthrough events (ABEs), characterized in rare instances by hypophysitis, colitis, and pancreatitis, they have been seen mainly in what are thought to be immunologically driven malignancies such as melanoma and renal cell carcinoma. Clinical trials are currently underway in patients with metastatic prostate cancer.
CTLA-4 is the prototype of checkpoint blockade, but there are other molecules that may offer additional targets for blockade. There are at least seven members of the extended B7 family of molecules, and at least three of these-B7-H1, B7-H3, and B7x (the latter identified by Allison's group)-also inhibit T-cell response but do so at later stages than CTLA-4.
Cytokines and Whole-Cell Vaccines: The GVAX Approach
Early vaccine trials made use of manipulated autologous tumor-cell vaccines. It soon became clear, however, that something more than the introduction of these cells was needed. Allogeneic tumor cells or cell lines were considered, but concern arose about human leukocyte antigen (HLA) histocompatibility and the possibility of lack of T-cell recognition in the absence of a recognized compatible HLA haplotype. Even if a short-lived antitumor response were feasible, it was unclear how the antitumor effect could be better sustained, ie, how to keep the immune-generated cells at the tumor site to maintain the antitumor effect.
Gene-transfer technology has enabled the enhancement of tumor-cell immunogenicity when used in vaccines. This was facilitated by the introduction of cytokine genes into cell lines with the idea of having the tumor cell act as the immunogen or stimulator and actively secrete a factor that would favor recruitment of other effector cells systemically. The cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) was shown by Dranoff et al[42] to provide potent antitumor immunity when transduced into B16 melanoma cells, a weakly immunogenic tumor cell line. Compared with other immune-modulating cytokines, GM-CSF provided the most specific, potent, and durable antitumor responses. This was due to the recruitment of granulocytes, macrophages, and antigen-presenting cells (APCs) such as dendritic cells, otherwise known as Langerhans cells, within the skin. The recruitment of APCs suggested that GM-CSF could create an environment ripe for antigen presentation.[26]
Simons et al[27,28] initially studied the immune response preclinically in animals injected with either LNCaP or PC-3 prostate cancer cell lines transduced with the gene for the production of GM-CSF. Simons' team[30] extended this approach further in a phase I clinical trial by inserting the GM-CSF gene into prostate cancer cells derived from individual patients' prostatectomy specimens. Autologous prostate cancer cells were established as primary cultures, transduced with the replication-defective retrovirus containing cDNA-encoding GM-CSF (MFG-GM-CSF), as described previously.[27-29]
These genetically modified prostate cancer cells (GVAX) were then lethally irradiated and assessed for GM-CSF secretion and MFG-GM-CSF integration. This "vaccine" was administered intradermally as two injections of 0.5 X 107 cells every 21 days until the patient's vaccine cell repository was exhausted. Vaccine site skin biopsies showed infiltrates of dendritic cells and macrophages. Biopsy of skin sites showing a delayed-type hypersensitivity response revealed infiltrates of effector cells consisting of CD45 RO+ T cells and degranulating eosinophils consistent with activation of both Th1 and Th2 T-cell responses. A unique kind of vasculitis was also seen near the autologous tumor cells at the vaccine site. This trial demonstrated not only the feasibility of this approach, but also that it was safe and could induce both humoral and T-cell responses against prostate cancer cell-associated antigens.
This proof of principle has led to the further development of this technology in more advanced-phase trials. GVAX now uses allogeneic whole tumor cells from the established prostate cancer cell lines LNCaP and PC-3 in lieu of autologous tumor cells derived from the patient. These cell lines were selected based on their complementary behavioral patterns, which represent the basic dynamics of prostate cancer growth: LNCaP is a hormone-sensitive line expressing a number of differentiation antigens including PSA and PSMA, with a mutant androgen receptor similar to that found in prostate tumors. PC3 represents a diametrically opposed aggressive cell line that is not hormone-sensitive and is suggestive of an aggressive phenotype. It does not express PSA, but rather, expresses urine plasminogen activator, metalloproteinases, and neuroendocrine peptides, all of which are suggest a hormone-resistant state.
Clinical Trials
To date, five clinical trials with GVAX prostate cancer vaccine have been conducted in more that 200 patients with recurrent or hormone-resistant prostate cancer.[30] In an early trial of 34 patients with asymptomatic metastatic hormone-refractory prostate cancer (HRPC), the median survival of patients treated with the vaccine was 26.2 months. This is consistent with findings in patients treated with standard-of-care chemotherapy.
Another study enrolled 80 patients who received the vaccine at 200 X 106 cells monthly, 500 X 106 cells biweekly, or 500 X 106 cells as a priming dose followed by 300 X 106 cells biweekly. Of 19 patients in the high-dose group, 6 (32%) had PSA declines, albeit not immediately but after several months of vaccinations. Almost all patients had an immune response irrespective of dose level, although the greatest percentage of patients with an immunoglobulin (Ig)G response against the tumor cell line was in the highest dose level. The vaccine was also safe and thus far has not produced any dose-limiting toxicities.
Currently, two trials are ongoing in the United States with survival as the primary endpoint, VITAL-1 is for patients without cancer-related pain and compares GVAX vs docetaxel and prednisone. A second trial in prostate cancer, VITAL-2, is for patients with cancer-related pain and compares GVAX plus docetaxel vs docetaxel and prednisone. Additional objectives include the proportion of patients who develop skeletal-related events, progression of disease, and time to onset of bone pain. Recent data presented by Gerritsen et al[43] suggest that an antitumor effect can be induced by combining GVAX with an immune modulating agent.
A recent phase I trial studied the combination of GVAX and MDX-010, a humanized monoclonal antibody that blocks the inhibitory effects of CTLA-4. When the agents were given together, patients experienced regression of bone metastases. Interestingly, a decline in PSA seemed to correspond with the development of autoimmune hypophysitis manifested as either adrenal insufficiency or autoimmune thyroiditis. The development of these conditions seemed to portend a more favorable response to treatment in a manner often seen with melanoma patients who develop vitiligo following immune treatment with vaccine. Overall, the combination was safe. Skin biopsies confirmed inflammatory infiltrates. A phase III trial of GVAX and anti-CTLA-4 monoclonal antibody in patients with castrate metastatic prostate cancer is wider development.
How to Channel a Minimal Immune Response for Maximal Gain
The harnessing of dendritic cells pulsed with specific peptides has led to several phase I/II trials in patients with hormone-resistant prostate cancer. Thomas-Kaskel et al[25] conducted a phase I/II trial of dendritic cells pulsed with prostate stem cell antigen (PSCA) and PSA peptides on 12 HLA-A2+ patients with advanced prostate cancer. Cellular immune response against the vaccine was the primary endpoint. The vaccine proved safe, with one patient exhibiting complete regression of lymph node disease despite rising PSAs, and four patients showing stable disease. While the median survival of all patients was 13.4 months, a significantly superior survival (P = .003) was noted in patients with delayed-type hypersensitivity responses to the antigens.
Another approach that targets both humoral cellular components of the immune response uses autologous leukapheresis cellular products that are incubated with a novel immunogen-a recombinant fusion protein consisting of prostatic acid phosphatase (PAP) linked to GM-CSF. Here, too, GM-CSF plays a critical role in the induction and recruitment of immune cells. This cellular product vaccine was another proof of principle utilizing a novel protein to stimulate the immune system. Preclinical studies in rats were directed at eliciting prostate cancer-specific immunity, ie, breaking immune tolerance. This was done ex vivo by loading purified dendritic cells (the most potent antigen-presenting cells) with an engineered antigen-cytokine fusion protein (PA2024) consisting of PAP and GM-CSF.[44] Delay of tumor development and improved survival were observed in tumor prevention models. Dendritic cells pulsed with PAP alone generated inferior immune responses.
This approach was brought to the clinical arena as an immunotherapy product (APC8015, now known as sipuleucel-T [Provenge]), which consists of autologous dendritic cells loaded ex vivo with the PAP/GM-CSF recombinant fusion protein. Patients underwent leukapheresis to remove mononuclear cells without using any mobilizing cytokine. The product was immediately sent to the Dendreon Corporation (Mountain View, Calif) for processing; dendritic cell precursors were collected and purified, then incubated with the fusion protein PA2024. The CD54+ cells were then returned to the patient within 24 hours and reinfused within 8 hours of formulation.
Sequential phase I and II trials in patients with hormone-resistant prostate cancer have been performed.[44] A total of 31 patients were treated; 12 with castrate metastatic disease were treated in the phase I portion, and 19 who were hormone-resistant but had no radiographic evidence of disease were treated in the phase II portion. The immune product was found to be safe, with the most common side effect being a brief febrile reaction during the cell product reinfusion. Overall, no improvement in bone scans or soft-tissue disease was seen in the phase I trial. The median time to disease progression for the phase I patients was 12 weeks, and the median time to progression for the phase II patients was 29 weeks. Among the 19 phase II patients, disease had not progressed in 7 by the end of the planned 1-year follow-up. Cytokine production by T cells responding to the target antigen was assessed, with the cytokine profile revealing that T cells released interferon-gamma but not IL-4. This suggested that the T-cell responses were more consistent with Th-1-type responses usually associated with host immunity to tumors.
These results established the framework for a recently reported inaugural phase III multicenter double-blind placebo-controlled trial randomizing patients to treatment with sipuleucel-T or placebo.[45] This trial was designed to test the effect of sipuleucel-T on time to progression and survival in patients with HRPC. A total of 127 patients with asymptomatic metastatic HRPC were randomized in a two-to-one ratio to receive three infusions of sipuleucel-T or placebo every 2 weeks. The trial included a crossover to the treatment arm of sipuleucel-T if placebo patients developed disease progression. As seen in earlier trials, the treatment was safe, with infusion-associated rigors followed by pyrexia being the most common side effects.
As early as 8 weeks posttreatment, the median ratio of the T-cell stimulation index-a measure of immunologic recognition of the immune product-was approximately eightfold higher in the sipuleucel-T arm vs placebo (P < .001). A total of 115 patients experienced disease progression. The study did not demonstrate an improvement in the primary endpoint of time to progression in patients treated with sipuleucel-T (Figure 2A).[45] Historically, the median time to progression has been about 3 months. However, in an intent-to-treat analysis, the current study demonstrated a 4.5-month improvement in overall survival (P = .01, Figure 2B).


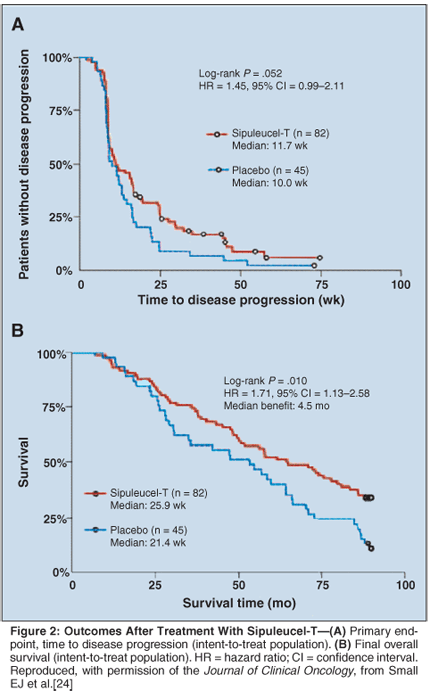
This seminal trial is the first phase III study to suggest that an immune strategy could lead to a modest improvement in overall survival. Other confirmatory trials are ongoing, but the nature of the effector population involved in eliciting the antitumor effects remains unclear, as does the best way to standardize the minimum number of effector cells needed for treatment.
Future Directions
There is renewed enthusiasm for immune therapies in the treatment of early and late relapsed prostate cancer. Current trends favor the use of multiple immune strategies, which are leading not only to our better understanding of how the immune system functions, but how it responds to new drug challenges. To this end, clinical trials with combinations of vaccine and docetaxel have been shown to be safe without inhibiting specific T-cell responses.[46] Interestingly, the addition of an immune modulator such as anti-CTLA-4 after cancer vaccine failure is under consideration as a salvage strategy.[47] Continued efforts to enhance immune responses remain an active research interest.
Disclosures:
The author has no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
1. James EL: Vaccination against cancer may result from discovery by British medical experts. The Globe, no. 23,566, July 17, 1925.
2. Ghosh H, Heston WDW: Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem 91:528-539, 2004.
3. Schulke N, Varlamova OA, Donovan GP, et al: The homodimer of prostate specific membrane antigen is a functional target for cancer therapy. Proc Natl Acad Sci USA 100:12590-12595, 2003.
4. Davis MI, Bennett MJ, Thomas LM, et al: Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci USA 102:5981-5986, 2005.
5. Silver DA, Pellicer I, Fair WR, et al: Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 3:81-85, 1997.
6. Chang SS, Reuter VE, Heston WD, et al: Metastatic renal cell carcinoma neovasculature expresses prostate specific membrane antigen. Urology 57:801-805, 2001.
7. Chang SS, Reuter VE, Heston WD, et al: Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res 59:3192-3198, 1999.
8. Slovin SF: PSMA vaccines: Naked DNA and protein approaches. Clin Prostate Cancer 4:1-6, 2005.
9. Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, et al: In vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res 60:5237-5243, 2000.
10. Vallabhajosula S, Goldsmith SJ, Kostakoglu L, et al: Radioimmunotherapy of prostate cancer using 90Y- and 177Lu-labeled J591 monoclonal antibodies: Effect of multiple treatments on myelotoxicity. Clin Cancer Res 11:7195s-7200s, 2005.
11. Divgi CR, Bander NH, Scott AM, et al: Phase I/II radioimmunotherapy trial with iodine-131-labeled monoclonal antibody G250 in metastatic renal cell carcinoma. Clin Cancer Res 4:2729-3279, 1998.
12. Milowsky MI, Nanus DM, Kostakoglu L, et al: Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J Clin Onc 22:2522-2531, 2004.
13. Morris MJ, Divgi CR, Pandit-Taskar N, et al: Pilot trial of unlabeled and indium-111-labeled anti-prostate specific membrane antigen antibody J591 for castrate metastatic prostate cancer. Clin Cancer Res 11:7454-7461, 2005.
14. Bander NH, Milowsky MI, Nanus DM, et al: Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Onc 23:4592-460, 2005.
15. Slovin SF: Immunologic targeting: How to channel a minimal response for maximal outcome. Curr Opin Urol 16:179-185, 2006.
16. Slovin, SF, Ragupathi G, Adluri S, et al: Carbohydrate vaccines in cancer: Immunogenicity of a fully synthetic globo hexasaccharide conjugate in man. Proc Natl Acad Sci USA 6:5710-5715, 1999.
17. Slovin SF, Ragupathi G, Musselli C, et al: Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: Clinical trial results with alpha-N-acetylgalactosamine-O-serine/threonine conjugate vaccine. J Clin Oncol 21:4292-4298, 2003.
18. Slovin SF, Ragupathi G, Fernandez C, et al: A bivalent conjugate vaccine in the treatment of biochemically relapsed prostate cancer: A study of glycosylated MUC-2-KLH and Globo H-KLH conjugate vaccines given with the new semi-synthetic saponin immunological adjuvant GPI-0100 or QS-21. Vaccine 23:3114-3122, 2005.
19. Slovin, SF, Ragupathi G, Fernandez C, et al: Thomsen-Friedenreich (TF) antigen as a target for prostate cancer vaccine: Clinical trial results with TF cluster (c)-KLH plus QS21 conjugate vaccine in patients with biochemically relapsed prostate cancer. Cancer Immunol Immunother 54:694-702, 2005.
20. Arlen PM, Gulley JL, Todd N, et al: Antiandrogen, vaccine and combination therapy in patients with nonmetastatic hormone refractory prostate cancer. J Urol 174:539-546, 2005.
21. Gulley J, Chen AP, Dahut W, et al: Phase I study of a vaccine using recombinant vaccinia virus expressing PSA (rV-PSA) in patients with metastatic androgen-independent prostate cancer. Prostate 53:109-117, 2002.
22. Elzey BD, Siemens DR, Ratliff TL, et al: Immunization with type 5 adenovirus recombinant for a tumor antigen in combination with recombinant canarypox virus (ALVAC) cytokine gene delivery induces destruction of established prostate tumors. Int J Cancer 94:842-849, 2001.
23. Johnson LE, Frye TP, Arnot AR, et al: Safety and immunological efficacy of a prostate cancer plasmid DNA vaccine encoding prostatic acid phosphatase (PAP). Vaccine 24:293-303, 2006.
24. Scher HI, Eisenberger M, D'Amico AV, et al: Eligibility and outcomes reporting guidelines for clinical trials for patients in the state of a rising PSA: Recommendations from the Prostate-Specific Antigen Working Group. J Clin Onc 22:537-556, 2004.
25. Thomas-Kaskel A-K, Zeiser R, Jochim R, et al: Vaccination of advanced prostate cancer patients with PSCA and PSA peptide-loaded dendritic cells induces DTH responses that correlate with superior overall survival. Int J Cancer 119:2428-2434, 2006.
26. Eager R, Nemunaitis J: GM-CSF gene-transduced tumor vaccines. Mol Ther 12:18-27, 2005.
27. Simons JW, Mikhak B, Chang J-F, et al: Induction of immunity to prostate cancer antigens: Results of a clinical trial of vaccination with irradiated autologous prostate tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor using ex vivo gene transfer. Cancer Res 59:5160-5168, 1999.
28. Simons JW, Jaffee EM, Weber CE, et al: Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte-macrophage colony-stimulating factor gene transfer. Cancer Res 57:1537-1546, 1997.
29. Berns AJ, Clift S, Cohen LK, et al: Phase I study of non-replicating autologous tumor cell injections using cells prepared with or without GM-CSF gene transduction in patients with metastatic renal cell carcinoma. Hum Gene Ther 6:347-368, 1995.
30. Simons JW, Carducci MA, Mikhak B, et al: Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naive prostate cancer. Clin Cancer Res 12:3394-3401, 2006.
31. Leach DR, Krummel MF, Allison JP, et al: Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734-1736, 1996.
32. Chambers CA, Kuhns MS, Allison JP: Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci USA 96:8603-8608, 1999.
33. Kwon ED, Foster BA, Hurwitz AA, et al: Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proc Natl Acad Sci USA 96:15074-1507, 1999.
34. Maker AV, Phan GQ, Attia P, et al: Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: A phase I/II study. Ann Surg Oncol 12:1005-1016, 2005.
35. Blansfield JA, Beck KE, Tran K, et al: Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 28:593-598, 2005.
36. Attia P, Phan GQ, Maker AV, et al: Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 23:6043-6053, 2005.
37. Hurwitz AA, Sullivan TJ, Sobel RA, et al: Cytotoxic T lymphocyte antigen 4 (CTLA-4) limits the expansion of encephalitogenic T cells in experimental autoimmune encephalomyelitis (EAE)-resistant BALB/c mice. Proc Natl Acad Sci USA 99:3013-3017, 2002.
38. van Elsas A, Hurwitz AA, Allison JP: Combination immunotherapy of B16 melanoma using anti-CTLA-4 and GM-CSF producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190:355-366, 1999.
39. van Elsas A, Sutmuller RP, Hurwitz AA: Elucidating the autoimmune and anti-tumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 (CTLA-4) blockade in combination with a B16 melanoma vaccine: Comparison of prophylaxis and therapy. J Exp Med 194:481-489, 2001.
40. Camacho LH, Ribas A, Glaspy JA, et al: Phase 1 clinical trial of anti-CTLA4 human monoclonal antibody CP-675,206 in patients (pts) with advanced solid malignancies (abstract 2505). Proc Am Soc Clin Oncol 22:164s, 2004.
41. Jaeger MA, Stroehlein A, Schoberth A, et al: Immunotherapy with the trifunctional antibody removab leads to significant elimination of tumor cells from malignant ascites in ovarian cancer: Results of a phase I/II study (abstract 2504). Proc Am Soc Clin Oncol:164, 2004.
42. Dranoff G, Jaffee E, Lazenby A, et al: Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA 90:3539-3543, 1993.
43. Gerritsen W, Van Den Eertwegh AJ, De Gruijl T, et al: A dose-escalation trial of GM-CSF-gene transduced allogeneic prostate cancer cellular immunotherapy in combination with a fully human anti-CTLA antibody (MDX-010, ipilimumab) in patient with hormone-refractory prostate cancer (mHRPC) (abstract 2500). Proc Am Soc Clin Onc 24:100s, 2006.
44. Small EJ, Fratesi O, Reese DM, et al: Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 18:3894-3903, 2000.
45. Small EJ, Schellhammer PF, Higano CS, et al: Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metstatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 24:3089-3094, 2006.
46. Arlen PM, Guiley JL, Parker C, et al: A randomized phase II study of concurrent docetaxel plus vaccine versus vaccine alone in metastatic androgen-independent prostate cancer. Clin Cancer Res 12:1260-1269, 2006.
47. O'Mahony D, Morris JC, Quinn C, et al: A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin Cancer Res 13:958-964, 2007.












