Heterogeneity and Cancer
Cancer heterogeneity, long recognized as an important clinical determinant of patient outcomes, was poorly understood at a molecular level. Genomic studies have significantly improved our understanding of heterogeneity, and have pointed to ways in which heterogeneity might be understood and defeated for therapeutic effect.


Figure 1: Cancers Can Be Heterogeneous at Many Different Levels
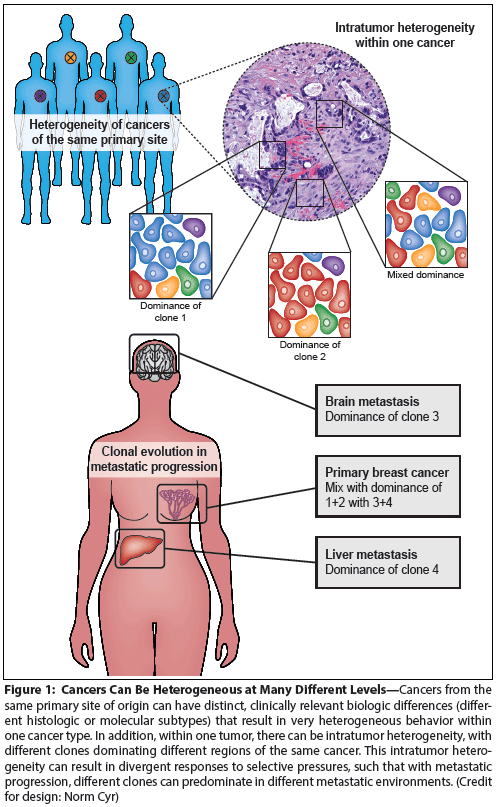


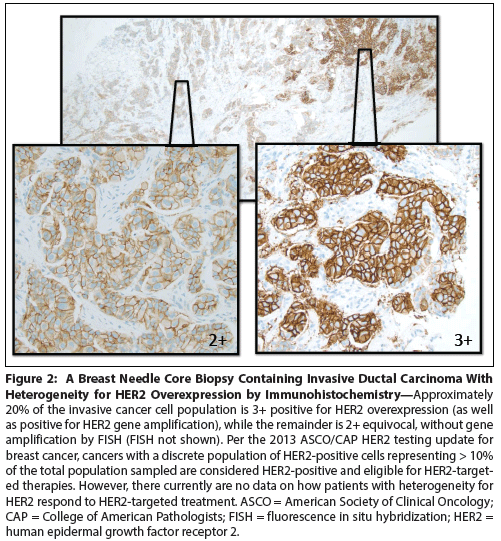
Figure 2: A Breast Needle Core Biopsy Containing Invasive Ductal Carcinoma With Heterogeneity for HER2 Overexpression by Immunohistochemistry
Cancer heterogeneity, long recognized as an important clinical determinant of patient outcomes, was poorly understood at a molecular level. Genomic studies have significantly improved our understanding of heterogeneity, and have pointed to ways in which heterogeneity might be understood and defeated for therapeutic effect. Recent studies have evaluated intratumoral heterogeneity within the primary tumor, as well as heterogeneity observed between primary and metastasis. The existence of clonal heterogeneity in the primary and metastasis also affects response to therapy, since the Darwinian pressures of systemic therapy result in clonal selection for initially rare variants. Novel technologies (such as measurements of circulating tumor cells and circulating tumor DNA) may allow physicians to monitor the emergence of clonal subtypes and intervene at an early point to improve patient prognosis.
Introduction
Cancer is a heterogeneous disease. Practically from the moment pathologists first looked at human cancers under the microscope, they saw that differing histologic appearances could define distinct subtypes of cancers from the same primary site of origin. These histology-based definitions of cancer subtypes have been modified and refined over time to elucidate both prognosis and prediction of response to specific treatments. Molecular data have revealed how radically different cancers from one primary site can be, and using this information, we have refined and in many cases revolutionized cancer classification systems. Oncologists can now use knowledge of the intertumor heterogeneity of each cancer type to treat patients with more personalized and targeted therapies that lead to better outcomes.
However, heterogeneity in cancer is not limited to differences between different patients, but also occurs within a single patient (Figure 1). This intrapatient or intratumoral heterogeneity can present great challenges for cancer treatment. When oncologists first began to treat patients with systemic therapy, they noticed that some metastatic deposits grew while others shrank. Such mixed responses still occur, suggesting that intratumoral heterogeneity is a living force that follows classic Darwinian patterns. However, it is still currently unclear what level of heterogeneity for a drug target is required to thwart the drug’s successful elimination of the cancer.
Studies of intratumoral heterogeneity have proliferated in recent years. These studies have developed from two converging sources. First, recent years have seen the creation of numerous agents that target specific biologic pathways, each with its own particular resistance mechanisms. Secondly, the “omics” revolution has revealed the true nature of intratumoral heterogeneity at multiple levels-and such epigenomic, genomic, and proteomic analyses have enriched our understanding of why cancers grow and kill.
Doctors and patients deal with tumor heterogeneity on a daily basis. A pathologist notes that some tumor cells in a breast cancer are amplified for human epidermal growth factor receptor 2 (HER2) by fluorescence in situ hybridization (FISH), while others express normal HER2 copy numbers. A medical oncologist, treating a patient with metastatic colorectal cancer with epidermal growth factor receptor (EGFR)-targeted therapy, observes growth of one liver metastasis while another shrinks.
This heterogeneity at the molecular, cellular, tissue, and organ levels bedevils clinical cancer care because it allows cancers to evolve and evade available therapeutics. Wherever significant heterogeneity exists, it serves as a potential threat to the life of the patient. However, we currently understand very little about what type of heterogeneity is the most threatening.
What have we learned regarding tumor heterogeneity in recent years? What are its sources and how does it manifest itself? How does it affect the natural history of human cancers-and their unnatural history in the face of cancer therapeutics? This paper will discuss recent observations derived from molecular and clinical/translational studies that help elucidate the mystery of tumor heterogeneity.
Defining Clinically Relevant Heterogeneity
Some level of heterogeneity exists in all cancers, but defining what the most clinically relevant cell populations are in a cancer will depend heavily on its definition and on the setting of critical thresholds. Immunohistochemical assays of HER2 have suggested the existence of heterogeneity in anywhere from < 1% to 30% of tumors, although a carefully performed recent analysis suggests a rate of 5% for FISH.[1] However, the frequency of a positive result will depend on both the definition of a single “positive cell,” as well as the threshold set for the percentage of positive cells in the population that will qualify the result as positive.
What percentage of cells needs to be positive for a specific biomarker or mutation to consider the cancer “positive” and likely to respond to a specific therapy? Clearly a breast cancer with 1% estrogen receptor (ER) positivity is not likely to respond to hormonal therapy to the same extent as one with greater than 95% of cells positive, yet both are considered ER-positive breast cancers and candidates for hormonal therapy. The heterogeneity present in the low ER expresser is clinically relevant and will make it more likely that additional therapeutic modalities will be required for successful treatment. The original study by Harvey et al[2] examining the relationship between ER expression levels and response to hormonal therapy showed the best disease-free survival (DFS) curves for patients with the highest levels of ER expression (Allred scores of 7–8). However, because the patients with mid- to low-level ER expression (Allred scores of 3–5) still had better DFS than those with < 1% or no expression (Allred scores of < 2), it was recommended that the threshold for offering treatment be an Allred score of 3 or greater (corresponding to at least 1% of cells with weak staining). While few studies have been able to demonstrate that the relative degree of benefit of hormonal therapy is in a direct or linear relationship to the level of hormone receptor expression, current American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) hormone receptor testing guidelines for breast cancer recommend the positive threshold of 1% of cells weakly positive but also recommend as standard the reporting of the percentage and intensity of cells staining positive (not just a positive or negative result).[3]
The distribution of heterogeneous cell populations may be clinically significant as well. For HER2 testing by in situ hybridization (ISH), amplified cells can be present diffusely (as in the classic HER2-amplified breast cancer) or as a minor population in intermixed or clustered patterns.[4,5] Although there are limited data to suggest significant differences in outcomes between clustered and intermixed minority HER2-amplified cells, the 2013 ASCO/CAP HER2 testing guideline update for breast cancer recommends counting clustered cell populations separately.[4] As Figure 2 illustrates, these clustered HER2-positive minority populations may be evident on immunohistochemistry and missed on ISH testing unless the entire area is hybridized and scanned under the fluorescent microscope. Interestingly, the majority of breast cancers with results in the equivocal range or close to the threshold for positivity have the intermixed form of HER2 heterogeneity, still considered to be of uncertain clinical significance.[5]
Sometimes heterogeneity for the same biomarker can have different implications in different cancer types. Heterogeneity for HER2 gene amplification is more common in gastroesophageal and gastric cancers than in breast cancers.[6-8] Therefore, current recommendations for HER2 testing of gastric cancers (based on results of the ToGA trial) have been modified from the ASCO/CAP guidelines on HER2 testing in breast cancer to allow for smaller clusters of positive cells to be interpreted as a positive result in gastric biopsies.[4,9]
Ideally, the definitions and thresholds of these heterogeneous markers will be clinically validated so they can optimally predict outcomes and treatment benefit. However, because borderline cases are often not a large proportion of clinical trial populations, we are often left to use limited evidence or nonclinical data to make decisions about these definitions. In addition, it is possible that for some drug targets, heterogeneity may not be as clinically significant as for others. For example, the death of the targeted population might elicit a host immune response such that newly exposed antigens could result in an immune attack on the remainder of the cancer cells.
The Clinical Face of Intratumoral Heterogeneity
Intrapatient tumor heterogeneity presents multiple clinical challenges, from the initial diagnosis to the treatment of metastatic disease. At diagnosis, a cancer treatment plan is made, based largely on the pathology results from a single tissue sample of a primary site. While this method of sampling may be representative and sufficient for characterization of many early-stage cancers, it may under-sample cancers that are more heterogeneous, such that potentially druggable targets are not identified early on. With metastatic progression, heterogeneity can be even more pronounced, raising the question of which lesion to sample in order to identify the most useful treatment targets.
In the setting of treatment for metastatic disease, the presence of a mixed response (simultaneous growth and shrinkage of metastatic deposits) has long been recognized and is due to heterogeneous disease. We may underestimate key differences between metastatic lesions using current schema; eg, current Response Evaluation Criteria in Solid Tumors (RECIST) sum tumor measurements, so that both patients experiencing a partial response and those with progressive disease may have individual tumors whose vector is opposite to that of the overall measurement.
In the largest study to date evaluating such mixed responses, van Kessel and colleagues evaluated 290 patients enrolled in the CAIRO and CAIRO 2 colorectal cancer studies for the presence of mixed response.[10] Overall, 25 of 290 patients (8.6%) had a true mixed response, defined as having 2 lesions showing progression vs response. If one adds in patients with > 30% difference in individual lesion response, an additional 25.2% of patients could be said to have had a mixed response. Mixed responses are statistically common. They are also clinically relevant: Looking at patients with a partial response by RECIST criteria, patients with a mixed response had significantly worse survival than patients with homogenous partial responses (median overall survival of 23.7 vs 36.0 months, respectively; P = .019). Heterogeneity kills.
Molecular Heterogeneity in Primary Tumors
Recent large-scale deep sequencing efforts have shed light on the degree of molecular heterogeneity seen in primary tumors across the broad spectrum of human cancers. A study of 12 tumor types by investigators with The Cancer Genome Atlas (TCGA) project revealed a total of 127 significantly mutated (ie, driver-mutated) genes, with 2 to 6 drivers on average per individual.[11] Some of these mutated driver genes (particularly transcriptional factors/regulators) tend to be tissue-specific, and some (such as histone modifiers) are relatively more ubiquitous.
Mutational pairings are common in the TCGA data set, with 148 co-occurring pairs. Some pairings are disease-specific (mutated IDH1 and ATRX in glioblastoma, mutated TBX3 and MLL4 in lung adenocarcinoma), but most are not. Similarly, some mutations are tumor-specific (eg, mutations in NPM1 and FLT3 in acute myeloid leukemia[AML]), but most are not.
Lawrence et al examined genomic heterogeneity in 3,083 tumor-normal pairs across 27 tumor types.[12] Their analysis, published in Nature last year, focused not on driver mutations per se, but on overall somatic mutation frequencies. The median frequency of nonsynonymous mutations varied by more than three orders of magnitude across the cancer spectrum.
Approximately half of this variation can be explained by tissue type, with the highest mutational frequency occurring in highly mutagenized tissues (eg, skin/melanoma, lung). But even within a specific tumor type, there may be substantial genomic variability, some of which reflects underlying etiology (eg, head and neck squamous cell cancer, where virally induced cancers have fewer mutations than those that are tobacco-induced).
Etiology may also affect mutational heterogeneity, as Lawrence et al report.[12] For instance, the mutations in lung cancers are dominated by C→A mutations resulting from exposure to polycyclic aromatic hydrocarbons in tobacco smoke, while melanomas have frequent C→T mutations caused by misrepair of ultraviolet-induced covalent bonds between adjacent pyrimidines. Such etiology-related intertumor heterogeneity across cancer types is unsurprising; more interesting is the role of etiology in the development of heterogeneity within a tumor type. Stransky et al demonstrated that for head and neck squamous cell cancers, there is a significant difference between those caused by tobacco and those that are induced by human papilloma virus (HPV).[13] In contrast to HPV-induced cancers, tobacco-induced cancers carry a higher overall mutational load, and are far more likely to harbor p53 mutations. Such etiology-based intertumor heterogeneity significantly affects outcome.
Large-scale sequencing efforts by TCGA and related organizations almost certainly underestimate the degree of intratumoral heterogeneity. Gerlinger et al recently studied intratumoral heterogeneity in 10 patients with clear-cell renal cell carcinoma in whom multiple regions of the tumor were analyzed.[14] Significant intratumoral molecular heterogeneity was seen in all 10 tumors, with three-quarters of all driver mutations identified as being subclonal rather than ubiquitous. In the words of the authors, genomic analysis based on a single biopsy specimen creates “an illusion of clonal dominance.”
At an even deeper level, genomic analyses of single cells within a tumor have revealed an astonishing degree of heterogeneity. Wang et al performed deep sequencing of ~50 single cancer cell nuclei in two breast cancers, one ER-positive and one triple-negative, and demonstrated the existence of three classes of mutations: clonal mutations, found in a population sample and the majority of single cells; subclonal mutations, found in two or more cells; and de novo mutations (primarily point mutations), found in single cells.[15] Subclonal mutations were quite common, similar in frequency to clonal mutations. The authors concluded that, in the cells examined, “No two single tumor cells are genetically identical.” Heterogeneity is universal at the cell level.
Heterogeneity Between Primary and Metastasis
The intratumoral heterogeneity seen across a range of cancers also manifests itself in comparisons of primary and metastatic lesions. Gerlinger et al have given us a unifying approach to viewing such mutations based on an analysis of primary renal clear-cell tumors and their metastases.[16] Some mutational events are ubiquitous, appearing across all tumors; some mutational events occur across all metastases; and some are “private” mutational events limited to specific metastases. One possible outcome of this approach would be to focus therapeutically on so-called “truncal” mutations (those that occur in all primary and metastatic sites) on the assumption that these are both important and more likely to result in global responses. Similar results have recently been reported in melanoma, where 82% of patients have a gene expression pattern of “private” mutations among different metastatic sites.[17]
There is a growing literature examining concordance between primary and metastatic cancers for a variety of growth factor receptors, including ER and HER2 in breast cancer, EGFR in colorectal cancer and adenocarcinoma of the lung, and BRAF in melanoma. Knowledge of concordance is clinically valuable: Either loss of function in a pre-existing receptor, or gain of a new or altered receptor, might have important clinical implications. However, such assessments have been fraught with difficulty: differential specimen handling, inadequate tissue, denatured tissue, use of different assays over time-all these have limited our ability to compare primary and metastatic samples.
Breast cancer is perhaps the best-studied disease in this regard, with well-performed prospective analyses for ER, progesterone receptor (PR), and HER2. As recently reviewed by Penault-Llorca et al, discordance is greater for steroid receptors (15% to 40%) than for HER2 (8% to 10%). HER2 gain (as measured by FISH) is more common than HER2 loss; both gain and loss are common for ER and PR, although in patients receiving adjuvant hormonal therapy, loss is more frequently seen.[18]
Targeted therapies clearly alter either growth factor receptors or resistance-related downstream pathway molecules across a variety of human cancers. In the case of treatment with targeted therapies, one sees clear evidence of clonal evolution in response to Darwinian pressures. Several examples of this tendency can be seen across a variety of tumors:
(1) ER mutations following endocrine therapy. Mutations in the gene encoding the ER are uncommon in primary breast cancers. In contrast, tumors treated with endocrine manipulations (particularly multiple lines of hormonal therapy) have an increased frequency of ligand-binding domain mutations that render them less sensitive to endocrine manipulation. Jeselsohn et al recently reported recurring somatic mutations in codons 537 and 538 within the receptor ligand-binding domain in 12% of patients with metastatic disease, increasing to 20% for heavily treated patients.[19] These results are in line with earlier suggestions that intratumoral heterogeneity for ER mutations plays an important role in hormone resistance.[20,21]
(2) Loss of ER following adjuvant hormonal therapy. A recent analysis of the larger literature for hormone receptor discrepancies between primary and metastasis conducted by Sighoko et al concluded that the majority (but not all) of the discrepancies seen between primary and metastatic disease are due to technical misclassification.[22] With correction for misclassification, discordance between primary and metastasis was 12.4%, with positive-to-negative changes (10.1%) outnumbering negative-to-positive changes (2.3%) by approximately 5:1 for ER.
(3) EGFR mutations in adenocarcinoma of the lung. Activating mutations in EGFR represent an important target in lung adenocarcinomas. Chen and colleagues examined intrapatient heterogeneity for EGFR mutation expression in patients with matched tumors.[23] In patients in whom multiple paired lung metastases were sampled, there was a discordance rate of 24.4%. In patients exposed to EGFR inhibition between two biopsies, the discordance rate was 26.3% (10 of 38 patients); overall, 29.4% of tyrosine kinase inhibitor (TKI)-resistant tumors were heterogeneous for TKI expression.
To what extent are the mutations seen in advanced disease already present in the initial cancer? Until recently, this question was difficult to address for technical reasons. The advent of techniques allowing for high coverage of specific mutations, as well as clever mathematical modeling, has delivered at least an initial answer.
Diaz et al studied this question in the context of acquired resistance to EGFR inhibition in advanced colorectal cancer.[24] In this setting, KRAS mutation represents the predominant mechanism of resistance. Examining serial serum samples from 28 patients who received the anti-EGFR monoclonal antibody panitumumab, they found that 9 of 24 initially KRAS wild-type tumors developed detectable KRAS mutations, with 3 having multiple different KRAS mutations. Mathematical modeling suggested that these KRAS mutations were present prior to initiation of therapy, and that “the time to recurrence is simply the interval required for the subclone to repopulate the lesion.”
Similarly, Watanabe et al examined KRAS mutations in primary and metastatic colorectal adenocarcinomas.[25] While overall concordance between primary and metastatic sites was high, 11.6% of cases were discordant. Microdissection revealed a heterogeneous KRAS mutational pattern in the primary tumor in all cases; sampling error provides a straightforward explanation for seeming discordance.
Might therapy itself alter tumor heterogeneity? Landau et al investigated the evolution and impact of subclonal mutations in chronic lymphocytic leukemia (CLL).[26] Measuring mutations at multiple time points in 18 CLL patients, they demonstrated clonal evolution in 10 of 12 chemotherapy-treated patients, but in only 1 of 6 nontreated patients. This evolution took the form of the addition of new driver mutations (and increases in measurable genetic diversity), with expansion over time.
Similarly, Ding et al have examined the fate of eight patients with AML, comparing prechemotherapy and post-relapse genomes.[27] Deep sequencing revealed two clonal evolution patterns: “(1) the founding clone in the primary tumor gained mutations and evolved into the relapse clone, or (2) a subclone of the founding clone survived initial therapy, gained additional mutations, and expanded at relapse.” A significant gain in transversions for relapse-specific mutations suggests that cytotoxic chemotherapy induced those mutations and contributed to the underlying heterogeneity associated with relapse.
Pfeifer et al measured BCR-ABL kinase domain mutations during imatinib treatment in patients with imatinib-naive and newly diagnosed or recurrent Philadelphia chromosome–positive acute lymphoblastic leukemia.[28] The use of highly sensitive ligation-mediated polymerase chain reaction (PCR) and denaturing high-performance liquid chromatography, rather than standard PCR techniques, allowed for greater sensitivity in detection of mutations. Within 4 weeks of initiation of therapy, 33% of treatment-naive patients and 70% of pretreated patients expressed tyrosine kinase mutations in peripheral blood samples, up from 17% and 42%, respectively, at baseline. The rapidity of this effect supported the idea that selection of pre-existing mutant clones rather than induction of new mutations was occurring in these patients.
Thus, the jury is still out on the frequency and importance of treatment-related heterogeneity. Darwinian pressures clearly select for pre-existent mutations, conferring a survival advantage in the face of therapy. At the same time, it would be totally unsurprising that therapy, particularly DNA-damaging chemotherapy and radiotherapy, might increase and create intratumoral heterogeneity.
Clinical Implications of Tumor Heterogeneity
As described above, the genomic landscape, like the clinical and pathologic landscapes that preceded it, is one where tumor heterogeneity is common and important. What are the implications for clinical practice at the current time, and how will our growing understanding of tumor heterogeneity affect future cancer care?
The demonstration that metastatic cancers, particularly those that have received an intervening targeted therapy, can differ in important ways from the primary tumor is the foremost and most easily exploitable finding. Intervening targeted therapy can have two obvious effects: gain of function and loss of function. Gain of function may involve the addition (or, more often, unveiling) of growth factor receptors. An example of this would be an ER-positive breast cancer, which, at the time of recurrence, overexpresses HER2, potentially rendering that cancer sensitive to an additional targeted therapy. Loss of function may be similarly important. In the case described above, a breast cancer could also lose ER activity, rendering the patient insensitive to further endocrine manipulations.
While retesting metastatic sites for growth factor receptors is logical to consider and prudent to perform, there are remarkably few data on the practical outcome of such retesting. If a patient previously documented as having a HER2-negative primary breast tumor now tests positive at a metastatic site, will her outcome be improved as a result of targeted therapy delivered based on retesting? There is no large database testing this simple hypothesis for any growth factor receptor in any cancer.
Nevertheless, monitoring heterogeneity of metastatic clones seems eminently rational. Hochhaus et al recently examined treatment-emergent mutations in patients randomly assigned to treatment with either imatinib or nilotinib for BCR-ABL–mutant chronic myelogenous leukemia (CML).[29] Frontline nilotinib was associated with fewer, and less diverse, BCR-ABL mutations in chronic-phase CML. It is reasonable to suggest, although it remains to be proven, that real-time monitoring for emergent mutations (ie, monitoring for expanding tumor heterogeneity) might lead to early interventions, reducing the rate of tumor progression. Of course, not all cancers share CML’s near-absolute dependence on a single growth pathway, so monitoring with therapeutic intent will not be as simple for more heavily mutagenized cancers. The development of circulating tumor DNA technology may assist in such monitoring.[30]
The recent discovery of ER mutations in patients previously treated with hormonal manipulations is an example of both the promise and challenge posed by such analyses. Should we never treat patients whose cancers harbor such mutations with hormonal therapies? Should they be automatic candidates for chemotherapy? We currently do not know the answer to such questions.
Intratumoral heterogeneity affects individual patients; intertumor heterogeneity affects the entire clinical research enterprise. One of the hallmarks of the past decade has been the steady application of targeted therapies to patients harboring specific mutations. Quite consistently, across a large variety of human cancers, such therapies have produced remissions that are initially profound but frighteningly brief. Analysis of the mutational landscape of breast cancer (to offer one example among many) in 100 breast cancers revealed driver mutations in 40 different cancer genes, found in 73 different combinations, with the number of driver mutations ranging from 1 to 6 per patient.[31] A simplified therapeutic approach utilizing monoform sequential targeted therapy will manifestly fail. Yet how will we ever be able to design trials targeting 73 different combinations for every 100 patients? Intertumor heterogeneity is a potential disaster for the clinical trials approach that has dominated the past 2 decades of research.
Thwarting Heterogeneity
Heterogeneity itself is a major issue for cancer therapy. Some tumors, as TCGA analyses demonstrate, are characterized by high mutational rates and seemingly endless heterogeneity. In these cases, the issue is not simply a qualitative one (“How do we shut off mutation X?”) but also a quantitative one (“How do we respond to mutations A, B, C…X, Y, Z?”). Indeed, in highly mutagenic tumors, the tendency is to give in to despair, for the kinase-based approaches that have dominated the last decade are unlikely to alter the outcome of highly heterogeneous/significantly mutated tumors.
We may discover, in the fairly near future, that there is some mutational load (expressed as significant heterogeneity) inconsistent with cure using kinase-based therapeutic approaches. If so, being able to recognize that we are about to hit a therapeutic “wall” might have the benefit of directing us down alternative therapeutic pathways. Certainly approaches such as mobilization of the immune system (eg, regulatory T cells, checkpoint inhibitors) represent reasonable approaches for such patients.
For somewhat less heterogeneous tumors, Gerlinger’s suggestion that “truncal” mutations represent a potentially valuable point of attack also seems an appropriate approach, with the caveat that such attacks may well be thwarted by clonal evolution and resulting heterogeneity. An interesting example of this problem comes from one of the first analyses of genomic heterogeneity in pancreatic cancer. Evaluating serial metastases from a pancreatic primary tumor by deep sequencing, Campbell et al demonstrated that the KRAS mutation present in the primary tumor (a wonderful potential target, given its near ubiquity in pancreatic primaries) was lost in one, though not all, of the daughter metastases.[32] How common a problem this might be is unknown, but it never pays to bet against the ingenuity of cancer.
Financial Disclosure:Dr. Sledge has received clinical trial support and an honorarium for a lecture from Genentech; he is a member of the Board of Directors of Syndax and a member of the Scientific Advisory Board of Symphogen. Dr. Allison has no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
References:
1. Perez EA, Press MF, Dueck AC, et al. Immunohistochemistry and fluorescence in situ hybridization assessment of HER2 in clinical trials of adjuvant therapy for breast cancer (NCCTG N9831, BCIRG 006, and BCIRG 005). Breast Cancer Res Treat. 2013;138:99-108.
2. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17:1474-81.
3. Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Path Lab Med. 2010;134:e48-72.
4. Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997-4013.
5. Allison KH, Dintzis SM, Schmidt RA. Frequency of HER2 heterogeneity by fluorescence in situ hybridization according to CAP expert panel recommendations: time for a new look at how to report heterogeneity. Am J Clin Pathol. 2011;136:864-71.
6. Hofmann M, Stoss O, Shi D, et al. Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology. 2008;52:797-805.
7. Albarello L, Pecciarini L, Doglioni C. HER2 testing in gastric cancer. Adv Anat Pathol. 2011;18:53-9.
8. Pirrelli M, Caruso M, Di Maggio M, et al. Are biopsy specimens predictive of HER2 status in gastric cancer patients? Digest Dis Sci. 2013;58:397-404.
9. Wolff A, Hammond M, Schwartz J, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118-45.
10. van Kessel CS, Samim M, Koopman M, et al. Radiological heterogeneity in response to chemotherapy is associated with poor survival in patients with colorectal liver metastases. Eur J Cancer. 2013;49:2486-93.
11. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-9.
12. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214-8.
13. Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157-60.
14. Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225-33.
15. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155-60.
16. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883-92.
17. Harbst K, Lauss M, Cirenajwis H, et al. Molecular and genetic diversity in the metastatic process of melanoma. J Pathol. 2014;233:39-50.
18. Penault-Llorca F, Coudry RA, Hanna WM, et al. Experts’ opinion: Recommendations for retesting breast cancer metastases for HER2 and hormone receptor status. Breast. 2013;22:200-2.
19. Jeselsohn R, Yelensky R, Buchwalter G, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757-67.
20. Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. 2010;16:2702-8.
21. Fuqua SA, Gu G, Rechoum Y. Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat. 2014;144:11-9.
22. Sighoko D, Liu J, Hou N, et al. Discordance in hormone receptor status among primary, metastatic, and second primary breast cancers: biological difference or misclassification? Oncologist. 2014;19:592-601.
23. Chen ZY, Zhong WZ, Zhang XC, et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist. 2012;17:978-85.
24. Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537-40.
25. Watanabe T, Kobunai T, Yamamoto Y, et al.5Heterogeneity of KRAS status may explain the subset of discordant KRAS status between primary and metastatic colorectal cancer. Dis Colon Rectum. 2011;54:1170-8.
26. Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714-26.
27. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506-10.
28. Pfeifer H, Lange T, Wystub S, et al. Prevalence and dynamics of bcr-abl kinase domain mutations during imatinib treatment differ in patients with newly diagnosed and recurrent bcr-abl positive acute lymphoblastic leukemia. Leukemia. 2012;26:1475-81.
29. Hochhaus A, Saglio G, Larson RA, et al. Nilotinib is associated with a reduced incidence of BCR-ABL mutations vs imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood. 2013;121:3703-8.
30. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548-54.
31. Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400-4.
32. Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109-13.













