Metastatic Cancer in Solid Tumors and Clinical Outcome: Skeletal-Related Events
More than 50% of patients with advanced breast or prostate cancer have identifiable bone metastasis, and 30% to 40% of patients with non–small-cell lung cancer ultimately develop metastases to bone.[1,2]
Metastasis to the bone represents a frequent complication of visceral cancers, most commonly in patients with advanced breast, prostate, and lung cancer. More than 50% of patients with advanced breast or prostate cancer have identifiable bone metastasis, and 30% to 40% of patients with non–small-cell lung cancer ultimately develop metastases to bone. Most tumors preferentially metastasize to the axial skeleton, targeting the vertebrae, pelvis, proximal ends of long bones, and skull. Skeletal complications as a result of these metastases are widely recognized to increase mortality and decrease quality of life-specifically the loss of mobility, independence, and social functioning of a patient. Advances in understanding the mechanisms of metastasis to bone, the resulting physiologic disturbances that take place, screening, diagnosis, and availability of better treatment options are advancing clinicians’ abilities to combat this devastating problem.
More than 50% of patients with advanced breast or prostate cancer have identifiable bone metastasis, and 30% to 40% of patients with non–small-cell lung cancer ultimately develop metastases to bone.[1,2] Most tumors preferentially metastasize to the axial skeleton, targeting the vertebrae, pelvis, proximal ends of long bones, and skull.[3] Skeletal complications as a result of these metastases are widely recognized to increase mortality and decrease quality of life-specifically the loss of mobility, independence, and social functioning of a patient.
Bone has several features that render it a target of circulating tumor cells. Stephen Paget first noted this in 1889 in his “seed and soil” hypothesis, whereby he postulated that the complimentary characteristics of a target organ and circulating tumor cells determines where tumors ultimately metastasize.[4,5] With bone, the high blood flow to the marrow serves to harbor and supply certain tumor cells with the necessary media in which to grow and multiply. Successful metastasis is largely dependent on immediate survival during migration, clonal expansion, and development of local blood supply. Additionally, tumor cells express adhesive molecules that allow them to bind to stromal and matrix elements of the bone.
Healthy bone undergoes constant remodeling, directed by the influence of endocrine hormones like parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (1,24-(OH)2D3), paracrine hormones, and cytokines. In the setting of metastasis, the structural integrity of bone is compromised due to disruptions in the normal balance of osteolytic and osteoblastic activity. The resulting lesions leave the bone weakened and prone to damage even under low-impact stress events. This commonly results in debilitating bone pain and can also lead to other skeletal-related events (SREs) such as pathologic fractures, bone deformation, leukoerythroblastic anemia, subsequent surgery or radiation, threatening hypercalcemia of malignancy, and spinal cord or nerve root compression.[6] Most primary research, however, reports on four of these SREs: radiation, surgery, fractures, and spinal cord compression.
Incidence of SREs and Subsequent SREs
A recent retrospective review found the incidence of bony metastasis to be 24% and 30%, respectively in American and Japanese patient populations with non–small-cell lung cancer (NSCLC). Of these 66% were noted to have skeletal lesions at the time of their initial staging on positron-emission tomography (PET)/computed tomography (CT) scan.[7]
FIGURE 1


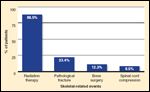
Distribution of skeletal-related events in 1-year retrospective study of a cohort of men with advanced prostate cancer metastasized to bone. Adapted from Lage et al [10].
In the placebo arm of a randomized control trial, Rosen et al found that patients with lung carcinoma or other visceral tumors with metastasis to bone (excluding carcinomas of the breast and prostate) experience an average of 2.71 SREs/year.[8] The risk of additional SREs after the first also increases with each event. Other studies have noted that patients can experience up to four SREs per year, the cumulative incidence of which can have negative consequences on overall survival.[9] One analysis of insurance claims of patients with metastatic prostate cancer found that 22% of patients experienced more than one SRE with a mean time of 2.9 months between identification of the first type and the second type of SRE (if they were different, eg, surgery and pathologic fracture).[10] This same study also found that most patients experience radiation therapy, as shown in Figure 1, when they experience a SRE. Only 23.4% of SREs were found to be pathological fractures.
RANK Ligand Pathway
TABLE 1


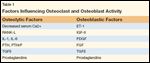
Factors Influencing Osteoclast and Osteoblast Activity
The increased risk of SREs in the setting of bony metastasis stems from a series of perturbations in the normal physiologic activity of bone remodeling. Normal bone is composed of two types: cortical bone, composed of hard, mineralized matrix; and trabecular or cancellous bone, which is less dense and more metabolically active. Cortical bone accounts for 85% of the total bone mass and can be found in the long bones of the appendicular skeleton. The other 15% makes up the cancellous bone that houses the red marrow, which contains stromal cells and hematopoietic stem cells that differentiate into osteoblasts and osteoclasts, respectively. Cancellous bone is found primarily in the vertebral bodies and pelvis. Osteoblasts are responsible for the deposition of new mineral matrix, creating new bone. Osteoclasts are responsible for the resorption of old bone in response to hormonal milieu. The mineralized matrix of bone contains immobilized cytokines and other growth factors that can directly influence neighboring cells-factors like tumor growth factor–beta (TGF-beta), insulin-like growth factor–2 (IGF-2), bone morphogenic proteins (BMPs), fibroblast growth factors (FGFs), and platelet-derived growth factor (PDGF).[11] Until bone is resorbed through osteoclastic activity, though, these cytokines remain fixed and nearby cells are not influenced by their hidden presence. A list of factors that regulate remodeling of bone is shown in Table 1.
The human body regulates calcium homeostasis and phosphate homeostasis in large part through parathyroid hormone (PTH), 1,25-dihydroxyvitamin D, and their action on bone, the GI tract, and the nephron. Calcitonin, a product of thyroid parafollicular C-cells, plays a small role in the activation of osteoblasts. In the bone, while osteoclasts oppose the actions of osteoblasts, the former cells, in fact, rely on the latter for cues in activity. Namely, osteoblast stimulation results in the release of receptor activator of RANK ligand. RANK ligand acts on its receptor, RANK, found on osteoclastic progenitor cells, to induce osteoclast formation and activity. At the same time, osteoblasts secrete osteoprotegerin (OPG), a decoy receptor of RANK ligand, effectively competing with RANK ligand to activate osteoclastic progenitor cells. The ratio of available RANK ligand and OPG, which is further influenced by local cytokines and growth factors, determines the net deposition or resorption of bone.
FIGURE 2


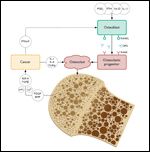
This depicts elements of normal bone physiology, namely the input of parathyroid hormone, 1,25-dihydroxyvitamin D, prostoglandins and cytokines in stimulation of osteoblasts to upregulate the expression of RANKL and release of osteoprotegerin (OPG), decoy receptors for RANKL that inhibit RANKL activity of corresponding receptors on osteoclastic progenitor cells. Osteoclasts will routinely resorb bone, releasing growth factors like IFG-II, TFG, PDGF, BMPs that serve to restore homeostatic balance by stimulating bone deposition and remodeling. With a metastatic lesion, the cancer, begins a “vicious cycle” by secreting factors like PTHrP and other cytokines to stimulate an osteolytic lesion, effectively creating a positive feedback loop that leads to more destruction and weakening of bone. Calcium and other byproducts of the destruction of bone mineral matrix, like urinary N-telopeptide (uNTx) are also released in the process of osteoclastic activity.
A so-called “vicious cycle” is created when tumor cells synthesize and release osteolytic factors, including PTH-related protein (PTHrP), interleukin-6 (IL-6), and tumor necrosis factor–alpha (TNF-alpha), among many others. These stimulate osteoclasts, which in turn resorb bone and then release the fixed growth factors, which positively influence the growth of tumor cells. These growth factors close a positive feedback loop on the tumor cells by activating them, creating an endless loop of activity that drives an osteolytic lesion.[12,13] These pathways and their pathologic deviations are depicted in Figure 2 and discussed in greater detail in the paper by Murthy et al earlier in this supplement.
Types of Bone Metastasis
Metastatic lesions of the bone can be classified in one of three ways: osteolytic, osteoblastic, or mixed.[14] Most cancers result in osteolytic lesions (carcinoma of breast and lung). In vivo studies have shown that osteolysis is due to osteoclastic activity, and is not a direct effect of the cancer itself. Overall, osteolytic lesions result from greater osteoclast than osteoblast cellular activity, resulting in uncoupled bone resorption. That is, there is no negative feedback mechanism to halt the process.[15] Targeting the various intersections within the positive feedback loop with appropriate therapies can clinically prevent further degradation of the bone, preserving function and preventing or delaying SREs.[16]
Prostate cancer (and 15% to 20% of breast cancers) results in osteoblastic lesions of the bone. In these lesions, the balance has been thought to shift to more bone deposition than destruction, as shown with correlation of levels of alkaline phosphatase and osteocalcin, both markers of osteoblastic activity. However, the bone that is deposited is of poor quality and often leads to severe bone pain and still puts the patient at risk for pathologic fractures and other subsequent SREs.
Research implicates the activity of Endothelin-1 (ET-1) as a mediator of this activity through both paracrine and autocrine activity on osteoblast cells. Prostate cancer and some breast cancer cells secrete high levels of ET-1, which in turn stimulate osteoblast cells.[17] An in vitro analysis found that an ET-1 receptor antagonist reduced osteoblastic activity.[18] The microenvironment of bone may further enhance ET-1 activity through the stimulation of prostate cancer cell proliferation and potentiating the effect of other local growth factors.[19]
That there exist two types of bony metastatic lesions is likely an oversimplification, as normal physiology dictates a carefully choreographed balance of activity from osteoclasts and osteoblasts. Perturbation of this healthy model does not likely inactivate one aspect of normal bone homeostasis, but rather results in an over-preponderance of bone deposition or destruction. This view was validated by a mouse model examining the activity of osteoblastic xenograft breast cancers.[20]
Researchers initially saw an increase in osteolytic activity followed by a resurgence in osteoblastic deposition of bone. Similarly, osteolytic lesions often result in new (albeit ineffective) bone deposition in response to the loss of healthy bone. As such, even in osteoblastic lesions specific to prostate cancer, osteolytic activity is comparable to that observed in breast cancer and multiple myeloma. Because osteolysis plays an important role in both lesions, it is believed that pharmacologic targeting-especially by bisphosphonates, which are potent inhibitors of osteoclasts-can serve to mitigate the horrendous clinical effects of such metastatic lesions.
Bone Markers as a Predictor of SREs
Osteoclastic activity results in the destruction of mineral matrix, which is composed primarily of type I collagen. Several metabolic products are released as a result, including urinary N-telopeptide (uNTx). Greenspan et al studied the level of one such marker in the setting of men undergoing ADT for prostate cancer and demonstrated that patients with higher levels of these metabolic byproducts were predisposed to greater loss of BMD.[21] Of note, the markers were elevated up to six months in advance of the measured loss in BMD, which suggests that such markers could be used to screen patients at increased risk of experiencing SREs. The possibility of using laboratory tests to determine, in advance, which individuals will most benefit from preventative intervention is tantalizing. Another study showed that patients with high uNTx levels had a greater incidence of SREs compared with patients with low levels of uNTx. Those with low uNTx levels also had better median survival (8.2 months vs 3.2 months).[22] Future prospective studies validating the use of these clinical markers to stratify patients need to be performed.
Burden of SREs
Clinical and Economic Impact of Pathological Fracture
The continued medical care of individuals who experience SREs constitutes a sizable cost that is ripe for targeting with prevention strategies listed herein.[23] A one-year study of the incidence of SREs, including radiation therapy, pathologic fracture, surgery, or spinal cord compression found that half of patients with prostate cancer and bony metastases experienced at least one SRE with a mean cost of $12,469 per patient.[10]
Patients that experienced more than one SRE within the study year (22% of the study population) consumed a mean of $26,384 worth of health care, significantly higher than those who experienced just one event ($8,484). The study also noted that 87% of patients who experienced an SRE did so within one year of their initial diagnosis of bony metastatic disease. These costs are comparable to those of SREs in the setting of advanced lung cancers.[24] Prevention strategies, timely diagnosis, and more cost-effective interventions have the potential to ultimately reduce both the clinical and economic burden of SREs.
Risk Factors, Screening and Diagnosis
A number of different factors increase a patient’s risk for developing skeletal morbidities as a consequence of bony metastasis. Some cancers, regardless of location, auto-secrete PTHrP, which interacts with the PTH receptor in osteoblasts upregulating the synthesis of RANKL. This results in bone resorption, which weakens the bone and increases the likelihood of a SRE.
Patients should be assessed at initial diagnosis of their cancer and worked up for signs and symptoms of bone metastasis. Clinicians should be prepared to note any bone pain, recent fractures, and changes in height or posture. Further testing and imaging will be guided by the type of cancer, stage, and symptomatology.
TABLE 2



Secondary Contributors to SREs
Patients with advanced visceral cancers are at high risk for the development of metastatic lesions to the bone that can be devastating for a patient and reduce overall survival. Given the severe consequences, clinicians should be aware of the risk factors for bone loss, osteopenia, osteoporosis, and SREs and diligently screen patients to identify any potential secondary causes of bone loss. This evaluation should include a thorough history and physical to identify any prior SREs such as fracture or vertebral deformity, predisposing medications like glucocorticoids, or the presence of social factors such as alcoholism that would otherwise contribute to the overall picture of skeletal health. A complete list of secondary antecedents to loss of bone mineral density that might potentiate a SRE is shown in Table 2.
The recommendations for screening patients for bony metastasis vary with the type of cancer. Several imaging modalities are available to clinicians to screen patients. Currently, imaging constitutes the mainstay of screening for metastasis to the bone, usually in the form of plain radiographs, magnetic resonance imaging (MRI), CT, PET, or radionuclide bone scan (bone scintigraphy). The sensitivity and specificity of each modality varies with cancer type and burden of disease. Nuclear imaging studies (bone scan and PET) can use a variety of novel radiolabels to detect cancer lesions.[25] Laboratory testing of serum markers of bone turnover (e.g., alkaline phosphatase, osteocalcin) can guide screening but is rarely definitive. Nuclear medicine tests such as PET and bone scan work primarily in the setting of high bone turnover, as their radioactive tracers are most attracted to these areas of metabolic activity.
The radionuclide bone scan is the most sensitive modality for detecting bony metastasis from prostate cancer.[26] In the PSA era, screening for bony metastasis in the absence of symptoms is not seen as necessary given the shift toward earlier diagnosis and more indolent disease; metastasis at initial presentation is rare. However, patients with bone pain or advanced disease (PSA > 20 ng/mL, Gleason > 8, or locally-advanced disease on digital rectal exam) should undergo a radionuclide bone scan. Men on androgen deprivation therapy should also receive attention even in the absence of symptoms, as they are at high risk for osteopenia, osteoporosis, and subsequent SREs.
Current recommendations state that patients with lung cancer should undergo a PET scan during initial staging and workup to evaluate for bony metastases. Alternatively, if this is not available, a bone scan can be performed.
Treatment Trends
Hormone Therapies Androgen deprivation therapy (ADT), often consisting of luteinizing hormone– releasing hormone (LHRH) agonists, is the preferred first-line therapy for metastatic prostate cancer to the bone. Unfortunately, as alluded to earlier, ADT can also impact skeletal health and often precipitates osteopenia or even osteoporosis, increasing the likelihood of a pathologic fracture.[27] Prostate cancer eventually overcomes androgen blockade and becomes refractory to such therapies, often requiring other interventions.
With breast cancer, there are a number of hormonal therapy options that are often tailored to the expression of estrogen and progesterone receptors as detected by histological staining. If large numbers of these receptors are present, pre-menopausal women may benefit from selective estrogen receptor modulators (SERMs), which can selectively downregulate receptors in specific tissues with estrogen receptors, namely metastatic tumors, while sparing other tissues (like bone).[28] For postmenopausal women with advanced breast cancer, selective aromatase inhibitors (SAIs) can reduce estrogen levels by blocking the action of aromatase, the enzyme that converts peripheral circulating androgens to estrogens.[29] These have been shown to be superior to SERMs in preventing disease progression and increasing survival, but unfortunately also result in greater resorption of bone and increase fracture risk as compared to SERMs.[30,31]
A 2-year, placebo-controlled, multicenter trial in United States and Mexico investigated the clinical significance of a SERM with respect to prostate cancer patients on androgen-deprivation therapy, with a primary endpoint of incident monomorphic vertebral fractures.[32] Secondary endpoints include bone mineral density (BMD), lipid levels, breast pain, and hot flashes. Initial results demonstrate clinically significant decreases in the incidence of vertebral fracture and increases in BMD. While this group of patients does not necessarily have bony metastasis, the results may herald a future for this drug class in the setting of osteolytic/osteoblastic metastases to prevent SREs.
Bisphosphonates This class of drug is unique in that it targets the feedback loop created between the metastatic lesion and bone, thus preventing or delaying onset of SREs.[33,34] Bisphosphonates bind to the active site of resorption of bone, entering osteoclasts and inducing apoptosis, thus preventing further resorption.[35] The goals of bisphosphonate therapy include preventing new and recurrent SREs, palliating bone pain, reducing the need for other therapies, and mitigating further morbidity. The efficacy of this drug class in attaining these clinical goals has been investigated in multiple placebo-controlled, randomized control trials showing significant improvements in clinical outcomes.[8,36,37] The use of bisphosphonates in the treatment of debilitating bone pain reduces the need for NSAIDs and opioid analgesics, which can have unintended consequences for patients with advanced cancer.
Because this type of therapy is systemic and lacks the myelosuppressive side effects of chemotherapy and radiation, it can be used as an adjunct to such therapies, broadening its use. Some studies have even demonstrated synergistic effects between bisphosphonates and chemotherapies.[38,39] Despite being systemic, these drugs localize to bone surfaces with active resorption, potentiating its activity and therapeutic efficacy.[40]
External Beam Radiation and Radiopharmaceuticals The use of external beam radiation prophylactically has been shown to limit clinical consequences for individuals suffering from new bony metastases to weight-bearing bones. Additionally, radiation to affected bone has been shown to palliate intractable bone pain that often results from these metastases but is often underutilized by clinicians.[41] The optimum dose of radiation and timing of delivery has been a hotly contested area of research, but a meta-analysis showed no significant difference between the different schedules.[42] In the United States, a survey of radiation oncologists showed that most employ 30 Gy in ten fractions.[43]
Similar to the nuclear radiolabels used in bone scans and PET scans, there are bone-seeking radiopharmaceuticals that concentrate in areas of high bone turnover and can be used for therapeutic intervention in patients with metastatic disease. These drugs target Src family kinases.[44,45]
Surgery If a bone metastasis threatens a weight-bearing bone (eg, femur), surgical intervention should be considered to prevent fracture. Surgical therapy is often followed by external beam radiation, which is then followed by systemic therapy, such as appropriate chemotherapy or use of bisphosphonates to treat bone pain and prevent future SREs.
Conclusions
Skeletal-related events remain a large source of morbidity and shorter life expectancies, and cost the health care system countless dollars in lengths of stay and outpatient care. Identifying and mitigating risk factors, timely diagnoses, and appropriate interventions can reduce the untoward effects of SREs. Research continues to elaborate mechanisms of osteolytic and osteoblastic lesions that are amenable to pharmacologic targeting to halt the cycle of bone destruction and weakening. With studies showing correlation between increased levels of biochemical markers of bone turnover and increased likelihood of SREs, disease progression and death, it may be possible to select for patients that are at greatest risk of SREs and implement prevention strategies using the medical therapies discussed, leading to more cost-effective and safer care for patients with metastatic bone disease.
Financial Disclosure:Dr. Crawford serves on speakers bureaus for Ferring, Watson, AstraZeneca, GlaxoSmithKline, and Indevus, and advisory boards for Ferring and Indevus. Dr. Rove has no other significant financial interest or relationship with the manufacturers of any products or providers of any service mentioned in this article.
This article was conceived of and fully funded by Amgen, and Amgen provided background direction for the article.
References:
1. Mundy GR: Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat Rev Cancer 2:284–293, 2002.
2. Coleman RE: Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat Rev 27:165–176, 2001.
3. Galasko CSB: The anatomy and pathways of skeletal metastases, in Weiss L, Gilbert AH (eds): Bone Metastases, vol 6, pp 49–63. Boston, GK Hall, 1981.
4. Paget S: The distribution of secondary growths in cancer of the breast. Lancet 1:571–572, 1889.
5. Fidler IJ: The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nature Rev Cancer 3:453–458, 2003.
6. Coleman RE: Skeletal complications of malignancy. Cancer 80:1588–1594, 1997.
7. Kosteva J, Langer C: The changing landscape of the medical management of skeletal metastases in nonsmall cell lung cancer. Curr Opin Oncol 20:155–161, 2008.
8. Rosen LS, Gordon D, Tchekmedyian NS, et al: Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, phase III, double-blind, placebo-controlled trial. Cancer 100:2613–2621, 2004.
9. Saad F, Lipton A: Clinical benefits and considerations of bisphosphonate treatment in metastatic bone disease. Semin Oncol 34:S17–S23, 2007.
10. Lage MJ, Barber BL, Harrison DJ et al: The cost of treating skeletal-related events in patients with prostate cancer. Am J Manag Care 14:317–322, 2008.
11. Hauschka PV, Mavrakos AE, Iafrati MD, et al: Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-sepharose. J Biol Chem 261:12665–12674, 1986.
12. Guise TA, Mundy GR: Cancer and bone. Endocr Rev 19:18–54, 1998.
13. Yin JJ, Selander K, Chirgwin JM, et al: TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 103:197–206, 1999.
14. Roodman GD: Mechanisms of bone metastasis. N Engl J Med 350:1655–1664, 2004.
15. Kozlow W, Guise TA: Breast cancer metastasis to bone: Mechanisms of osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia 10:169–180, 2005.
16. Chirgwin JM, Guise TA: Molecular mechanisms of tumor-bone interactions in osteolytic metastases. Crit Rev Eukarot Gene Expr 10:159–178, 2000.
17. Mohammad KS, Guise TA: Mechanisms of osteoblastic metastases: Role of endothelin-1. Clin Orthop Relat Res 415(suppl):S67–74, 2003.
18. Yin JJ, Mohammad KS, Käkönen SM, et al: A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A 20:10954–10959, 2003.
19. Chiao JW, Moonga BS, Yang YM, et al: Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer 83:360–365, 2003.
20. Yi B, Williams PJ, Niewolna M, et al: PDGF in breast cancer promotes osteosclerotic bone metastases. J Bone Min Res 15(suppl):A1035, 2000.
21. Greenspan SL, Coates P, Sereika SM, et al: Bone loss after initiation of androgen deprivation therapy in patients with prostate cancer. J Clin Endocrinol Metab 90:6410–6417, 2005.
22. Brown JE, Cook RJ, Major P, et al: Bone turnover markers as predictors of skeletal complications in prostate cancer, lung cancer, and other solid tumors. J Natl Cancer Inst 97:59–69, 2005.
23. Oefelein MG, Ricchiuti V, Conrad W, et al: Skeletal fractures negatively correlate with overall survival in men with prostate cancer. J Urol 168:1005-1007, 2002.
24. Delea T, Langer C, McKiernan J, et al. The cost of treatment of skeletal-related events in patients with bone metastases from lung cancer. Oncology 67: 390–396, 2004.
25. Messiou C, Cook G, deSouza NM: Imaging metastatic bone disease from carcinoma of the prostate. Br J Cancer 101:1225–1232, 2009.
26. Terris MK, Klonecke AS, McDougall IR, et al: Utilization of bone scans in conjunction with prostate-specific antigen levels in the surveillance for recurrence of adenocarcinoma after radical prostatectomy. J Nucl Med 32:1713, 1991.
27. Smith MR, Finkelstein JS, McGovern FJ, et al: Changes in body composition during androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 87: 599–603, 2002.
28. Love RR, Mazess RB, Barden HS, et al: Effects of tamoxifen on bone mineral density in postmenopausal women with breast cancer. N Engl J Med 32: 852–856, 1992.
29. Buzdar A, Douma J, Davidson N, et al: Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 19:3357–3366, 2001.
30. Heshmati HM, Khosla S, Robins SP, et al: Role of low levels of endogenous estrogen in regulation of bone resorption in late postmenopausal women. J Bone Miner Res 17:172–178, 2002.
31. Howell A, Cuzick J, Baum M, et al: Results of the ATAC (arimidex, tamoxifen, alone or in combination) trial after completion of 5 years adjuvant treatment for breast cancer. Lancet 365: 60–62, 2005.
32. Smith MR, Morton RA, Wallace H, et al: A phase III randomized controlled trial of toremifene to prevent fractures and other adverse effects of androgen therapy in men with prostate cancer (abstract LB-241). Proceedings of the 99th Annual Meeting of the American Association for Cancer Research. San Diego; April 15, 2009.
33. Rosen LS, Gordon D, Kaminski M, et al: Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: A randomized, double-blind, multicenter, comparative trial. Cancer 98:1735–1744, 2003.
34. Saad F, Gleason DM, Murray R, et al: Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst 96:879–882, 2004.
35. Licata AA: Discovery, clinical development, and therapeutic uses of bisphosphonates. Ann Pharmacother 39:668–677, 2005.
36. Kohno N, Aogi K, Minami H, et al: Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J Clin Oncol 23:3314–3321, 2005.
37. Weinfurt KP, Anstrom KJ, Castel LD, et al: Effect of zoledronic acid on pain associated with bone metastasis in patients with prostate cancer. Ann Oncol 17:986–989, 2006.
38. Ozturk OH, Bozcuk H, Burgucu D, et al: Cisplatin cytotoxicity is enhanced with zoledronic acid in A549 lung cancer cell line: Preliminary results of an in vitro study. Cell Biol Int 31:1069–1071, 2007.
39. Bertelli G, Heouaine A, Arena G, et al: Weekly docetaxel and zoledronic acid every 4 weeks in hormone-refractory prostate cancer patients. Cancer Chemother Pharmacol 57:46–51, 2006.
40. Henry D, von Moos R, Vadhan-Raj S, et al: A double-blind, randomized study of denosumab versus zoledronic acid for the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. Eur J Cancer 7(suppl):12, 2009.
41. Samant R: How should we describe the benefits of palliative radiotherapy? Curr Oncol 13:230–234, 2006.
42. Wu JS, Wong R, Johnston M, et al: Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys 55:594–605, 2003.
43. Chow E, Danjoux C, Wong R, et al: Palliation of bone metastases: A survey of patterns of practice among Canadian radiation oncologists. Radiat Oncol 56:305–314, 2000.
44. Fizazi K, Beuzeboc P, Lumbroso J, et al: Phase II trial of consolidation docetaxel and samarium-153 in patients with bone metastases from castration-resistant prostate cancer. J Clin Oncol 27:2429–2435, 2009.
45. Fizazi K: The role of Src in prostate cancer. Ann Oncol 18:1765–1773, 2007.

")






Late Hepatic Recurrence From Granulosa Cell Tumor: A Case Report
Granulosa cell tumors exhibit late recurrence and rare hepatic metastasis, emphasizing the need for lifelong surveillance in affected patients.


