Testicular Cancer: Maintaining the High Cure Rate
The management of germ cell tumors has advanced dramatically,with cure rates approaching 90% to 95%. Treatment of stage I/Aseminomas generally includes orchiectomy and adjuvant radiotherapy.Treatment of stage I/A nonseminomatous germ cell tumors involvesorchiectomy followed by retroperitoneal lymph node dissection oractive surveillance. One of the major advances has been the introductionof cisplatin-based chemotherapy for metastatic disease and thedevelopment of a system of risk attribution. The logical managementof any patient with curable disease is to provide curative therapy andthen follow the patient in a structured manner, to diagnose and treatany complications in a timely manner.
ABSTRACT: The management of germ cell tumors has advanced dramatically, with cure rates approaching 90% to 95%. Treatment of stage I/A seminomas generally includes orchiectomy and adjuvant radiotherapy. Treatment of stage I/A nonseminomatous germ cell tumors involves orchiectomy followed by retroperitoneal lymph node dissection or active surveillance. One of the major advances has been the introduction of cisplatin-based chemotherapy for metastatic disease and the development of a system of risk attribution. The logical management of any patient with curable disease is to provide curative therapy and then follow the patient in a structured manner, to diagnose and treat any complications in a timely manner.
In the second half of the past century, dramatic improvements were made in the management of advanced cancers of the genitourinary tract, with particular progress in the management of germ cell tumors. The management of metastatic testicular germ cell tumors has become one of the paradigms of successful treatment, reflecting advances in chemotherapy and an improved understanding of the principles of tumor biology and of the importance of multidisciplinary management. Nevertheless, with increasing experience, we have come to recognize that these approaches to treatment have some flaws, and that we must be careful if we are to maintain the high cure rates that have been achieved.
TABLE 1



Steps to Successful Management of Testicular Cancer
An increased understanding of risk factors has allowed us to tailor our treatment to the level of risk. We have come to realize that some of our treatment strategies are associated with significant late effects, and we have attempted to avoid these by modifying some treatment approaches. However, such attempts to improve treatment outcomes have failed, largely because of a reduction in the initial cure rate. This serves to illustrate a critically important principle- when effective treatment is available, modifications must be introduced in a thoughtful and structured manner to ensure that there are no hidden costs associated with the innovations under consideration (Table 1).
Pathobiology
Most germ cell tumors arise from tissues derived from primordial cells that originate within the genital ridges and usually migrate in the midline to the testicles, and less frequently, to the retroperitoneum, mediastinum, and pineal region. These cells are capable of differentiating along two major histogenetic lines, forming seminomas and nonseminomatous germ cell tumors (NSGCT).[1,2] All of these tumors have a common germ cell origin and, in the testicles, arise from precursor cells described histologically as carcinoma in situ.[3] Less than 5% of testicular cancers are lymphomas and other non-germ cell tumors.[ 1,4] The ability to differentiate along different pathways is of particular importance with regard to late relapse (see below).
Seminomas
The most common type of seminoma is the classical variant, composed of uniform, round, or polygonal cells with abundant cytoplasm and a centrally placed nucleolus.[1] Less common spermatocytic and anaplastic variants have been described, although it should be noted that the anaplastic variant sometimes represents a misdiagnosed NSGCT.[5] Seminomas are characterized by the presence of syncytiotrophoblastic giant cells in about 30% of cases, and these may produce a marker protein, human chorionic gonadotropin (HCG).
We have identified a subgroup of seminomas, morphologically resembling a solid variant of yolk sac carcinoma that is associated with a worse prognosis when treated with conventional radiotherapy or standard chemotherapy regimens.[5] Studies are in progress to define whether the biochemical determinants of outcome identified in the molecular revolution may explain the differences in the natural history of these tumors- for example, aberrations in expression of c-kit, other oncogenes, or tumor-suppressor genes.
Nonseminomatous Germ Cell Tumors
The group of NSGCTs includes several histologic subtypes: embryonal carcinoma, mature and immature teratoma, endodermal sinus tumor, and choriocarcinoma.[1] Frequently, NSGCT may consist of elements of undifferentiated cancer, trophoblastic tissue, and varying components of somatic differentiation, such as cartilage, glandular tissue, or hair.[1,2] Another tumor marker, alpha-fetoprotein (AFP) is classically produced by endodermal sinus tumor, although it is also associated with embryonal carcinoma. The presence of AFP in the circulation signifies the presence of nonseminomatous elements, even if pure seminoma has been diagnosed in the primary tumor. This constitutes an indication to manage the patient for a nonseminomatous germ cell malignancy.
Common Features
Testicular seminomas and NSGCTs share many features in common[ 1,2]: (1) Both occur predominantly in males aged 18 to 35 years; (2) both usually follow an orderly pattern of spread, from the testis to the surrounding supportive tissues and/or up the spermatic cord, to regional and distant lymphatic channels, and sometimes to visceral sites via blood-borne metastasis; (3) both are characterized by the elaboration of tumor markers and by common etiologic associations, including a characteristic marker of the short arm of chromosome 12,[6,7] testicular maldescent, carcinoma in situ of the testis,[3] and a less clearly explained association with the syndrome of multiple atypical nevi[8]; (4) both histologic patterns have been associated with a susceptibility gene localized to chromosome Xq27[9]; and (5) both are highly sensitive to the effects of chemotherapy.[10,11]
However, important differences exist between testicular seminomas and NSGCT, including a somewhat older age range for patients with seminoma, a slightly higher prevalence of second primary tumors among males with seminoma, different patterns of metastasis, and a substantial radiosensitivity in seminoma vs marked radioresistance in NSGCTs. Although the data are relatively preliminary, it appears that the c-kit ligand is expressed more heavily in seminomas than in NSGCTs,[12,13] and this leads to the potential for an increased chance of clinical improvement in response to modulators of these ligands, such as imatinib mesylate (Gleevec) or more specific developmental compounds, when used for seminoma as opposed to NSGCT.
When presenting at extragonadal sites, the biology of these tumor types is less similar: The seminomatous tumors tend to retain sensitivity to treatment with chemotherapy or radiotherapy, and patients with these tumors are relatively easily cured. By contrast, the extragonadal NSGCTs are associated with much higher relapse rates and inferior cure rates.[14-16] The reasons for these differences are unknown, as the testicular and extragonadal tumors share virtually identical histologies and etiologic associations and similar patterns of spread.
Presentation
Primary testicular cancer commonly presents as a painless enlargement of the testis, often noted on self-examination.[17] There may be local pain,[18] which is often associated with hemorrhage within the tumor. Occasionally, a large tumor will drag on the spermatic cord, causing referred pain in the region of the flank, which, therefore, does not necessarily indicate the presence of retroperitoneal lymph node metastases. Unless the patient has previously undergone surgery for testicular maldescent or scrotal violation as part of the initial management of the primary tumor (a cardinal error), inguinal lymph nodes are usually not involved. Drainage of the testis is typically to the lymph nodes at the level of the ipsilateral renal hilum.[19]
Symptoms
The symptoms of metastatic germ cell tumors are protean and depend on the sites of involvement.[20,21] Most commonly, the retroperitoneal lymph nodes are involved early, which can be associated with lumbar backache or central/lower abdominal pain. Occasionally, renal colic is due to obstruction of the ureters by advanced lymph node metastases. Pulmonary metastases may be asymptomatic or, if extensive, may be associated with dyspnea, cough, hemoptysis, or chest pain. Brain metastases, although uncommon, may manifest as headache, confusion, dementia, or focal neurologic syndromes, and occasionally may be detected in the routine staging workup.[ 22,23] Liver or bone involvement is now distinctly uncommon at presentation but may be associated with relapse. When these features are seen at presentation, choriocarcinoma should be considered as the most likely histologic subtype. It should be kept in mind, however, that seminoma will sometimes metastasize to bone.
TABLE 2



Stage Classification of Testicular Cancer
Germ cell tumors may be associated with nonspecific or constitutional symptoms, including weakness, sweats, fevers, malaise, and asthenia, especially in the case of advanced disease. Gynecomastia may indicate the presence of testicular cancer, especially in patients with a dominant element of choriocarcinoma either in a primary tumor, metastatic deposits, or both.
Extragonadal germ cell tumors manifest symptoms similar to those described above, although they are more heavily influenced by the site of origin. For example, pineal germ cell tumors may be associated with headache, confusion, visual changes, stroke-like syndromes, and Parinaud's syndrome.[14] The presentation of retroperitoneal germ cell tumors is identical to that of testicular tumors with retroperitoneal metastases, with the exception of an obvious testicular primary. Mediastinal germ cell tumors are commonly associated with dyspnea, cough, chest, or back pain, and occasionally with superior vena cava syndrome. Mediastinal NSGCTs are commonly associated with metastases at presentation, and the symptoms reflect the sites of those metastases (Table 2).[24,25]
Monitoring Treatment
The efficacy of treatment is routinely monitored by clinical assessment, using physical examination and appropriate imaging studies. Plain radiographs and computerized axial tomographic (CT) scans are the standard modalities, with CT having almost completely replaced lymphangiography.[ 26] For brain metastases, magnetic resonance imaging (MRI) is superior to CT scanning, but the role of MRI in the assessment of other sites of systemic disease remains controversial. Although preliminary evidence supports the use of positronemission tomography (PET) in assessing the presence of viable cancer in residual deposits after chemotherapy for germ cell tumors,[27] this technology has not yet secured a defined place in management, given conflicting reports regarding specificity and sensitivity.
Tumor Markers
The circulating tumor markers AFP and HCG should also be measured as part of the initial diagnostic work-up and when monitoring therapy. The first specimens should be obtained prior to primary surgery. AFP has a normal half-life in the circulation of 5 to 7 days, and HCG has a half-life of 24 to 36 hours.[28] Prolongation of a circulating marker's half-life after orchiectomy usually denotes occult metastatic disease, and indicates the need for further investigation and treatment.[28]
In patients with metastatic disease, the absolute levels of tumor markers constitute independent prognostic determinants. During chemotherapy, there may be a transient release of markers from dying cancer cells, causing a transient elevation of blood levels before they begin to decline according to normal half-life gradients. Thus, serial measurements should be taken to determine whether the patient is responding adequately to treatment. If these repeated measurements are not taken during the period of chemotherapy, tumor marker release will not be identified and the half-life calculation will be incorrect, suggesting a prolonged tumor marker decline (incorrectly implying the presence of resistant disease).
Treatment of Stage I/A Disease
One of the most controversial issues in the management of germ cell tumors is the treatment of tumors without evidence of metastatic disease (stage I or A).[29] Traditionally, stage I seminomas were treated by inguinal orchiectomy and adjuvant radiotherapy (doses of around 35-40 Gy, later reduced to 25-30 Gy). With this approach, cure rates approached 90% to 95%, especially after the introduction of CT and gallium scanning as part of the diagnostic work-up. More recently, an approach involving close observation after inguinal orchiectomy, with serial measurement of tumor markers and repeated CT scans, has been implemented.
The paradigm for this approach was initially developed for NSGCTs (see below), but recent studies have suggested that it may be safely applied to the management of seminomas.[ 30-32] It is important to note that the pattern of relapse of seminoma may be more insidious than that of NSGCT, and relapses with a later time of onset have been documented in many series. Thus, most active surveillance protocols for stage I seminoma require close follow-up for at least 5 years and include continued abdominal CT scans. Seminoma is also less commonly associated with tumor marker production, and thus, radiologic imaging assumes greater importance.
Surgical Approach
For stage I/A NSGCT, the situation is also complex. Traditionally, such tumors have been managed by inguinal orchiectomy, followed by a radical retroperitoneal lymph node dissection. This approach has produced cure in more than 90% of cases, and has even afforded cure in some patients with histologic more extensive the involvement of the lymph nodes by cancer, the greater the likely need for chemotherapy. evidence of micrometastases, without the need for chemotherapy; ie, the more extensive the involvement of the lymph nodes by cancer, the greater the likely need for chemotherapy.
The major disadvantage of this surgical approach is the risk of surgical complications, including pulmonary emboli, hemorrhage, and perioperative pain, and the occurrence of incompetent ejaculation secondary to transection of retroperitoneal nerves. The potential for recurrence is predicated on the T stage (or extent of tumor within the testes and surrounding tissues), with adverse prognostic factors including local extension to the spermatic cord and surrounding structures, undifferentiated histology, and lymphatic-vascular invasion.[33,34] In addition, failure of tumor markers to normalize with appropriate half-life times is associated with an increased risk of relapse[33] and constitutes an indication for chemotherapy.
Active Surveillance
Studies from the Royal Marsden Hospital[11] and the Danish Testicular Cancer Group[35] have clearly shown that adjuvant radiotherapy does not prolong survival after orchiectomy for stage I/A NSGCT, and this modality is no longer a treatment option in this context. However, studies from the United Kingdom forged the development of the policy of active surveillance for stage I/A NSGCT.[36,31] This approach, predicated on the high cure rates among patients with good-risk metastatic disease, requires meticulous initial diagnostic work-up and the exclusion of patients with established lymph node or visceral, small-volume metastases.
For patients without major risk factors and with normal imaging tests, the relapse rate after orchiectomy alone is less than 25%. Thus, appropriate surveillance allows 75% of patients to be spared the need for adjuvant therapy (lymphadenectomy or radiotherapy) after orchiectomy.[37] However, a higher percentage of patients will potentially relapse following surveillance than after retroperitoneal lymph node dissection, and will thus be candidates for chemotherapy.[29]
The most important risk associated with this approach is lack of compliance by either: (1) a patient who defaults from regular follow-up without receiving the adjuvant therapy (eg, retroperitoneal lymph node dissection) that might have granted him a cure, or (2) a physician who is unaware of the necessary follow-up procedures, waits too long to assess the patient for the development of metastases, and thereby reduces the potential for cure.
TABLE 3



Active Surveillance Schedules for Stage I/A Nonseminomatous Germ Cell Tumors After Inguinal Orchiectomy
There is no optimal follow-up schedule.[38] However, successful approaches have all been predicated on meticulous initial diagnosis and work-up, and a structured approach to routine follow-up (with serial clinical assessment as well as serial assessment of tumor markers, chest radiographs, and CT scans). With time, less frequent CT scans are performed, but the need for frequent monitoring of tumor markers and chest x-rays continues. Table 3 illustrates schedules of active surveillance that have been associated with high cure rates.[32,38-40] The common feature of these schedules is meticulous and close follow-up for the first 2 to 3 years.
In centers of excellence, where clinicians are experienced in the conduct of surveillance programs and a clear policy of patient selection and follow-up has been defined, this approach appears to be safe, producing cure rates of greater than 90% to 95%.[29,32,38,39] Some patients develop relapses that prove to be intractable to therapy,[32,38] and rarely, patients develop complications of chemotherapy, such as leukemia.[ 41] Nevertheless, cure rates among patients treated with adjuvant retroperitoneal lymph node dissections or radiotherapy protocols are also less than 100%, and thus, active surveillance remains a reasonable option. As noted previously, implementation of this approach by an inexperienced clinician without a defined and validated plan of surveillance and follow-up is potentially dangerous.
High-Risk Patients
The appropriate management of patients with stage I/A disease and adverse risk factors is also controversial. Cullen et al suggested that adjuvant chemotherapy after orchiectomy is effective in preventing early relapse in patients with locally extensive tumors, involvement of the spermatic cord, and other high-risk factors.[42] The obvious problem with this approach is the introduction of early and late toxicity in patients who would not relapse in this context if they were simply observed. For example, we have shown that up to 50% of patients with high-risk stage I/A disease do not relapse if they are simply observed.[33,38] Therefore, for high-risk disease, in a highly monitored setting, close observation may be a reasonable approach in the hands of an experienced investigator.
Another appropriate option is retroperitoneal lymph node dissection, which can identify lymph node involvement, and thus, patients who are likely to require chemotherapy. In this context, the problem is that some patients with spermatic cord involvement or vascular invasion in the primary tumor will relapse in the lungs or other visceral sites without evidence of retroperitoneal lymph node metastasis, and therefore are not helped by the procedure. Ideally, a patient with high-risk stage I testicular cancer should be referred to a center of excellence for a second opinion before a final plan of management is determined.
Treatment of Small-Volume Lymph Node Metastases
When evidence of small-volume (≤ 5 cm) lymph node involvement (stage IIA or IIB, stage B) is found in seminoma, patients usually receive radiotherapy to the ipsilateral lymph nodes in the pelvis, with extension to the para-aortic chain (including the involved nodes).[43] Although the trend recently has been toward dose reduction, most clinicians use a relatively standard radiation dose of 30 to 35 Gy to ensure local tumor control. There is clear evidence that prophylactic mediastinal lymph node irradiation does not improve outcome and may, in fact, compromise any subsequent chemotherapy. The preliminary experience with doses of 20 to 25 Gy suggests no increase in the pattern of early relapse, but longer follow-up will be necessary to determine whether such modifications are truly safe.
In the case of NSGCT, optimal management of early, stage II disease is controversial, although there is general agreement that radiotherapy has no role. However, proponents of retroperitoneal lymph node dissection view the standard surgical approach as offering both diagnosis and definitive treatment and cite surgical cure rates of up to 50%, particularly in patients with only microscopic evidence of lymph node involvement.[44] With this approach, chemotherapy can be used to salvage most relapsing patients.[45] In many centers, adjuvant chemotherapy is administered routinely to patients with more than three to five involved nodes.
First-Line Chemotherapy
The alternative approach is to offer first-line cytotoxic chemotherapy to these patients, based on cure rates approaching 100% among those with small-volume metastases limited to the retroperitoneal nodes. The benefit of this approach is that it limits the morbidity associated with surgery, although this may be offset by the acute toxicity of chemotherapy and the potential for late complications. To date, no randomized trial has attempted to resolve this issue for small-volume stage II/B disease, and the decision is usually predicated on the biases of the clinician and the preferences of the patient.
Williams et al addressed this issue in a study of 195 patients with stage II NSGCT who were randomized to two cycles of adjuvant chemotherapy or observation with salvage chemotherapy at the time of relapse.[46] However, this study included patients with extensive (> 5 cm) retroperitoneal lymph node metastases. Of the 98 patients who were observed, nearly 50% relapsed; most were salvaged with chemotherapy, and 93 were alive and disease-free at the time of reporting. Of the 97 patients allocated to adjuvant chemotherapy, 6 relapsed (5 of whom had actually not received any chemotherapy before the recurrence), and 94 of the 97 survived. There was no statistically significant difference in outcome.[46]
Marker-Only Disease
Another controversial issue is management of patients with marker- only disease, ie, those without evidence of specific lymph node or other metastases but with persistent elevation of serum marker levels after orchiectomy. Retroperitoneal lymph node dissection will identify occult lymphatic involvement in some cases and achieve cure in up to 50%. Nevertheless, the other 50% of patients will eventually require chemotherapy. Analogous to small-volume nodal metastases, cure rates with cytotoxic chemotherapy are close to 100%, although there is a risk of additional late toxicity as compared with surgery alone. My own practice with marker-only disease has been to use initial combination chemotherapy, reserving postchemotherapy surgery for the occasional patient in whom lymph node enlargement is subsequently identified on CT scan.
Advanced Lymph Node Involvement and Distant Metastases
For patients with lymph node metastases measuring more than 5 cm in diameter (stage IIC or stage C disease) and for those with visceral metastases (eg, lung, liver, bone), the treatment of choice is systemic chemotherapy.[ 45] Up to 70% of patients have achieved cure with the use of early versions of the PVB regimen (cisplatin [Platinol], vinblastine, bleomycin [Blenoxane]).[10,36] Subsequently, a less toxic regimen, in which vinblastine was replaced by etoposide, was shown to be equivalent to the conventional PVB regimen.
With increased experience, it has become possible to distinguish prognostic categories that require more intensive front-line chemotherapy, based on inferior outcomes with standard cisplatin-based chemotherapy.[ 47-49] Adverse prognostic factors include greatly elevated serum tumor marker levels,[47,48,50,51,45] increased volume of disease,[48,49] higher numbers of pulmonary metastases,[ 51,52] and more sites of involvement.[ 48,50,51,45] In the latter instance, metastatic disease in the brain, liver, and possibly bone, is associated with a worse prognosis among those treated with standard PVB therapy.[51] Investigators at Memorial Sloan-Kettering Cancer Center have developed an algorithm-predicated on tumor marker levels and extent of disease[ 47]-with which to calculate prognosis and identify patients who require specific dose-intensive treatment strategies.[53]
IGCCCG Classification System
TABLE 4



International Germ Cell Cancer Collaborative Group Classification of Metastatic Disease
Most systems divide poor-risk germ cell tumors into similar prognostic groups, but a recent Intergroup collaboration by the International Germ Cell Cancer Collaborative Group (IGCCCG) has produced a common classification system to ensure uniform reporting of the results of treatment (Table 4).[54] This classification, derived from cumulative experience with 5,202 NSGCTs and 660 seminomas from 10 countries, has related the criteria listed in Table 4 to three prognostic groupings, based on 5-year survival: a good-prognosis group with a 91% 5- year survival, an intermediate-prognosis group with a 79% 5-year survival, and a poor-prognosis group with a 48% 5-year survival. It must be emphasized that cure is possible with aggressive multimodality therapy even for patients in the worst prognostic groups, such as those with brain metastases.[22,23]
For patients with good- or intermediate- risk disease, randomized trials have led to the replacement of vinblastine by etoposide in combination chemotherapy, with a consequent reduction in acute toxicity and a retention of the high cure rates.[55] However, recent studies have reported occasional cases of acute leukemia in patients who received etoposidecontaining regimens, and this policy will require further reassessment with the passage of time. Attempts to drop bleomycin from combinations in order to reduce toxicity have resulted in a reduced cure rate,[56-58] and this approach has been abandoned in most centers. However, investigators at Memorial Sloan-Kettering believe that bleomycin can be safely omitted, provided the dose of etoposide is maintained at 500 mg/m2 per course of treatment.[59]
Three cycles of PEB (cisplatin, etoposide, bleomycin) has been shown to be equivalent to four cycles with respect to cure rate, and is associated with less toxicity,[60] although the number of patients with intermediate- risk tumors in this study was not sufficient to exclude a substantial difference in survival. It will be important to maintain caution when developing new trials that have as their primary end point the reduction of side effects, because there is a simultaneous risk of also reducing cure rates. For example, in addition to the risk associated with deleting bleomycin, randomized trials have shown that cure rates decrease if cisplatin is replaced by carboplatin (Paraplatin) in combination chemotherapy for germ cell tumors.[61]
Poor-Risk and Previously Treated Metastatic Germ Cell Tumors
The interpretation of the literature regarding optimal management of poor-risk germ cell tumors has been complicated because of the lack of uniform classification criteria.[62] Frequently, different patient groups have been included in reports of the management of poor-risk disease, and the apparent improvements in outcome for one program have often only reflected the types of cases treated rather than a truly improved survival. In addition to selection factors for poor-risk disease, other selection criteria relate to performance status, age, and resources of patients treated in some poor-risk protocols. This issue applies particularly to nonrandomized phase I or II clinical trials.
For example, it was initially reported that PVeBV, a high-dose combination of cisplatin, etoposide (VePesid), bleomycin, and vinblastine, yielded unsurpassed objective response rates and apparently improved survival in patients with so-called poor-risk disease. However, a randomized trial that compared this regimen with a standard- dose regimen failed to show any evidence of a significant benefit from this approach.[63]
Several recent reports have suggested that high-dose therapy with autologous bone marrow transfusion or peripheral stem cell support may improve outcome in patients with truly poor-risk disease (consistent with the international consensus classification), and in particular in those with relapsed germ cell tumors after initial chemotherapy and those with mediastinal NSGCT.[64-67] However, more extensive information is necessary from randomized clinical trials before a definitive statement on this topic can be made.
One useful approach for patients with poor-risk disease was developed in the United Kingdom by Newlands et al.[68] It involved the use of rapid cycling, dose-intensive therapy (within the conventional dose range), with alternating delivery of POMB (cisplatin, vincristine [Oncovin], methotrexate, bleomycin) and ACE (actinomycin D [Cosmegen], cyclophosphamide, etoposide). A mature series of 339 patients included 92 with poor-prognosis disease according to IGCCCG criteria, and showed longterm survival in 75% of this group.[69]
Novel Cytotoxic Agents
Recently, the novel cytotoxic agents paclitaxel[70] and gemcitabine (Gemzar)[71,72] were shown to have activity in previously treated patients, and their utility is currently being investigated in a series of phase II and III ongoing clinical trials. Although there are no final data reflecting their activity in previously untreated cases, it appears that there may be an objective single-agent response rate of up to 20% among patients who previously received cisplatin- containing regimens, depending on whether they achieved initial remissions.
These novel agents have been incorporated into combination regimens. For example, Motzer et al documented an objective response rate of 77% for patients with relapsed germ cell tumors treated with secondline paclitaxel, ifosfamide (Ifex), and cisplatin.[73] In heavily treated patients, Hinton et al showed that the combination of paclitaxel plus gemcitabine yields a response rate of 20%, and occasionally produces long-term survival.[74] Most likely, these agents will form the basis of many future trials, especially with an emphasis on poor-risk previously untreated cases.
Investigational protocols have been developed that incorporate some of the concepts enumerated above. For example, rapid cycling of doseintensive regimens of paclitaxel/ ifosfamide and carboplatin/etoposide, which appear to yield sustained remissions beyond 30 months in approximately 50% of patients with an unfavorable prognosis for salvage response.[ 73] Although there is no uniform classification of unfavorable prognosis for salvage treatment, the following poor-risk factors have been proposed by investigators at Memorial Sloan-Kettering:
• extragonadal primary site
• progressive disease after an incomplete response to first-line platinum therapy
• poor response or lack of response to prior treatment with cisplatin-plusifosfamide- containing conventionaldose therapy.
Similar experience reported by a German collaborative group demonstrated a 30% long-term survival in relapsed and refractory patients who previously received treatment with cisplatin-based regimens.[75] In this study, conventional-dose salvage therapy with paclitaxel, ifosfamide, and cisplatin was followed by one cycle of high-dose chemotherapy with carboplatin, etoposide, and thiotepa. In most of these investigations, the respective merits of case selection,improved supportive care, and the role of high-dose therapy have not really been defined.
Chronic Treatment Toxicity and Routine Follow-up
TABLE 5


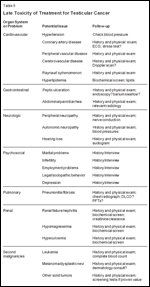
Late Toxicity of Treatment for Testicular Cancer
A detailed review of the toxicity of treatment is beyond the scope of this overview, and has been covered in detail elsewhere.[43,76,77] The acute toxicity of chemotherapy has been well defined, and includes the potential for nausea and vomiting, myelosuppression, alopecia, allergic phenomena, pneumonitis, infection, anorexia, and a range of relatively uncommon complications. Most of these can be controlled by modern supportive techniques.
The chronic or delayed side effects of treatment are increasingly recognized, especially as the medical community accepts germ cell tumors as a curable entity and the focus shifts to the avoidable costs of cure (Table 5).[40] Of particular concern is the emerging recognition of an apparent increase in the prevalence of cardiovascular and cerebrovascular disease, hypercholesterolemia, and a range of subtle metabolic abnormalities after cisplatin-based chemotherapy. Perhaps of greater concern is the prevalence of second malignancies, including leukemia, soft-tissue sarcoma, malignant melanoma, and other solid tumors.[76,78-80]
Because some of these disorders have been identified in surveys of patients conducted 5 to 10 years posttreatment, it is possible that the reported prevalence rates are low and will increase with the duration of follow-up, or that other unsuspected problems will emerge even later. As a consequence, the continuation of careful, focused follow-up will be essential for these patients, despite the efforts of many health maintenance organizations to reduce structured specialist follow-up by returning the care of these patients to their family practitioners.
Late Relapse
REFERENCE GUIDE
Therapeutic Agents
Mentioned in This Article
Actinomycin D (Cosmegen)
Bleomycin (Blenoxane)
Carboplatin (Paraplatin)
Cisplatin
Cyclophosphamide
(Cytoxan, Neosar)
Etoposide
Gemcitabine (Gemzar)
Ifosfamide (Ifex)
Imatinib mesylate (Gleevec)
Methotrexate
Paclitaxel
Thiotepa
Vinblastine
Vincristine
Brand names are listed in parentheses only if a drug is not available generically and is marketed as no more than two trademarked or registered products. More familiar alternative generic designations may also be included parenthetically.
Another problem of particular concern that has been identified increasingly in the past few years is the phenomenon of late relapse. Centers that manage large numbers of patients with germ cell tumors are beginning to see occasional patients with relapse at more than 10 years, characterized by malignant teratoma or the evolution of non-germ cell elements associated with germ cell tumors (including adenocarcinoma, soft-tissue sarcoma, or neuroendocrine carcinomas).
These conditions are proving to be difficult to treat effectively, and are associated with a high death rate. As yet, no optimal approach to treatment has been established. However, this phenomenon, accompanied by the evolving pattern of late toxicity, clearly illustrates the importance of ongoing follow-up in cancer centers for patients treated for germ cell tumors.
In general, our approach depends on the histology of the late relapse. Our management is usually multidisciplinary and particularly predicated on cytoreductive surgery and the use of purportedly non-cross-resistant chemotherapy. To this end, it appears that gemcitabine and the taxanes may be quite active in this context, especially for adenocarcinomas that arise from germ cell tumors. Because optimal management is not yet defined, we believe that such cases should be treated in tertiary referral centers if at all possible.
Overview
When considering the management of testicular cancer, a sense of perspective must be maintained. This disease was formerly a relentless killer of young men, and one must recognize that tremendous progress has been made in only 20 years. One should not make the mistake of modifying or delaying effective treatment to avoid the small risk of late complications. Instead, the logical approach is to offer curative therapy for patients with a curable cancer, and then to follow the patients in a structured fashion, allowing appropriate diagnosis and early management of any complications that ensue.
Financial Disclosure:The author has no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
1.
Mostofi FK, Price EB Jr: Tumors of theMale Genital System: Atlas of Tumor Pathology,2nd series, pp 1-175. Washington, DC,Armed Forces Institute of Pathology, 1979.
2.
Raghavan D, Neville AM: The biology oftesticular tumours, in Innes-Williams D,Chisholm G (eds): Scientific Foundations ofUrology, 2nd ed, pp 785-796. London, WilliamHeinemann Medical Books, 1992.
3.
Skakkebaek NE: Carcinoma in situ of thetestis: Frequency and relationship to invasivegerm cell tumours in infertile men. Histopathology2:157-170, 1978.
4.
Horwich A, Hamilton CR, Fisher C: Raretumours of the testis and paratesticular tissues,in Raghavan D, Brecher M, Johnson D,et al (eds): Textbook of Uncommon Cancer,2nd ed, pp 95-113. Chichester, NY, WileyLiss, 1998.
5.
Raghavan D, Sullivan AL, Peckham MJ,et al: Elevated serum alphafetoprotein and seminoma:Clinical evidence for a histologic continuum?Cancer 50:982-989, 1982.
6.
Atkin NB, Baker MC: i(12p): Specificchromosomal marker in seminoma and malignantteratoma of the testis? Cancer Genet Cytogenet10:199-204, 1983.
7.
Rodriguez E, Mathew S, Reuter V, et al:Cytogenetic analysis of 124 prospectively ascertainedmale germ cell tumors. Cancer Res52:2285-2291, 1992.
8.
Raghavan D, Zalcberg J, Grygiel JJ, et al:Multiple atypical nevi: A cutaneous marker ofgerm cell tumors. J Clin Oncol 12:2284-2287,1994.
9.
Rapley EA, Crockford GP, Teare D, et al:Localization to Xq27 of a susceptibility genefor testicular germ-cell tumours. Nat Genet24:197-200, 2000.
10.
Einhorn LH: Testicular cancer as a modelfor a curable neoplasm: The Richard andHinda Rosenthal Foundation Award Lecture.Cancer Res 41:3275-3280, 1981.
11.
Peckham MJ, Barrett A, McElwain TJ, etal: Non-seminoma germ cell tumours (malignantteratoma) of the testis: Results of treatmentand an analysis of prognostic factors. Br JUrol 53:162-172, 1981.
12.
Strohmeyer T, Reese D, Press M, et al:Expression of the c-kit proto-oncogene and itsligand stem cell factor (SCF) in normal andmalignant human testicular tissue. J Urol153:511-515, 1995.
13.
Bokemeyer C, Kuczyk MA, Dunn T:Expression of stem-cell factor and its receptorc-kit proto-oncogene in germ cell tumours. JPathol 122:301-306, 1996.
14.
Boyer MJ, Raghavan D: Extragonadalgerm cell tumors, in Peckham MJ, Pinedo H,Veronesi U (eds): Oxford Textbook of Oncology,pp 2169-2189. Oxford, Oxford UniversityPress, 1995.
15.
Shivdasani RA, Kantoff PW: Extragonadalgerm cell tumors, in Raghavan D,Scher HI, Leibel S, et al (eds): Principles andPractice of Genitourinary Oncology, pp 751-764. Philadelphia, Lippincott-Raven, 1997.
16.
Bokemeyer C, Nichols CR, Droz JP, etal: Extragonadal germ cell tumors of the mediastinum and retroperitoneum: Results from aninternational analysis. J Clin Oncol 20:1864-1873, 2002.
17.
Stephen RA: The clinical presentationof testicular tumours. Br J Urol 34:448-450,1962.
18.
Sandeman TF: Symptoms and early managementof germinal tumours of the testis. MedJ Aust 2:281-284, 1979.
19.
Donohue JP, Zachary JM, Maynard BR:Distribution of nodal metastases in nonseminomatoustestis cancer. J Urol 128:315-320,1982.
20.
Horwich A, Bajorin D: Testicular cancer:Presentation, assessment, and prognosis,in Raghavan D, Scher HI, Leibel S, et al (eds):Principles and Practice of Genitourinary Oncology,pp 671-682. Philadelphia, Lippincott-Raven, 1997.
21.
Bosl GJ, Motzer RJ: Testicular germcellcancer. N Engl J Med 337:242-253,1997.
22.
Raghavan D, Mackintosh JF, Fox RM, etal: Improved survival after brain metastases innon-seminomatous germ cell tumours withcombined modality treatment. Br J Urol 60:364-367, 1987.
23.
Bokemeyer C, Nowak P, Haupt A, et al:Treatment of brain metastases in patients withtesticular cancer. J Clin Oncol 15:1449-1454,1997.
24.
Horwith A, Bajorin D: Testicular cancer-presentation, assessment and prognosis,in Raghavan D, Scher HI, Leibel SA, et al(eds): Principles and Practice of GenitourinaryOncology, pp 671-682. Philadelphia, Lippincott-Raven, 1997.
25.
Greene FL, Page DL, Fleming ID, et al(eds): American Joint Committee on CancerStaging Manual, 6th ed, pp 317-322. NewYork, Springer, 2002.
26.
Hilton S, Schwartz LH: Radiologic imagingof genitourinary neoplasms, in RaghavanD, Scher HI, Leibel SA, et al (eds):Principles and Practice of Genitourinary Oncology,pp 33-42. Philadelphia, Lippincott-Raven, 1997.
27.
Stephens AW, Gonin R, Hutchins GD, etal: Positron emission tomography evaluationof residual radiographic abnormalities inpostchemotherapy germ cell tumor patients. JClin Oncol 14:1637-1641, 1996.
28.
Lange PH, Raghavan D: Clinical applicationsof tumor markers in testicular cancer,in Donohue JP (ed): Testis Tumors, pp 111-130. Baltimore, Williams & Wilkins, 1983.
29.
Jewett MAS, Lange PH, Raghavan D:Controversies in the treatment of early stagenonseminomatous testicular cancer, in RaghavanD, Scher HI, Leibel S, et al (eds): Principlesand Practice of Genitourinary Oncology,pp 703-713. Philadelphia, Lippincott-Raven,1997.
30.
Warde PR, Gospodarowicz MK, GoodmanPJ, et al: Results of a policy of surveillancein stage I testicular seminoma. Int J RadiatOncol Biol Phys 27:11-15, 1993.
31.
Francis R, Bower M, Brunstrom G, et al:Surveillance for stage I testicular germ cell tumours:Results and cost benefit analysis of managementoptions. Eur J Cancer 36:1925-1932,2000.
32.
Horwich A, Mason M, Hendry W: Testiculartumours, in Souhami RL, Tannock I,Hohenberger P, et al: Oxford Textbook ofOncology, 2nd edition, pp 2002-2035. NewYork, Oxford University Press, 2002.
33.
Raghavan D, Peckham MJ, HeydermanE, et al: Prognostic factors in clinical stage Inon-seminomatous germ-cell tumours of thetestis. Br J Cancer 45:167-173, 1982.
34.
Raghavan D, Vogelzang NJ, Bosl GJ,et al: Tumor classification and size in germcelltesticular cancer: Influence on the occurrenceof metastases. Cancer 50:1591-1595,1982.
35.
Rorth M, Jacobsen GK, von der MaaseH, et al: Surveillance alone versus radiotherapyafter orchiectomy for clinical stage I nonseminomatoustesticular cancer. J Clin Oncol9:1543-1548, 1991.
36.
Peckham MJ, Barrett A, Husband JE, etal: Orchiectomy alone in testicular stage I nonseminomatousgerm-cell tumours. Lancet2:678-680, 1982.
37.
Raghavan D: ‘Active surveillance’ forstage I testis cancer: Attaining maturity at 21years. Eur J Cancer 36:1891-1894, 2000.
38.
Raghavan D, Colls B, Levi J, et al: Surveillancefor stage I non-seminomatous germcell tumours of the testis: The optimal protocolhas not yet been defined. Br J Urol 61:522-526, 1988.
39.
Sturgeon JF, Jewett MA, Alison RE,et al: Surveillance after orchiectomy for patientswith clinical stage I nonseminomatoustestis tumors. J Clin Oncol 10:564-568,1992.
40.
Raghavan D: Follow-up of the patientwith testicular cancer, in Johnson FS, Virgo K(eds): Cancer Patient Follow-up. St. Louis,Mosby-Year Book, 1995.
41.
Segelov E, Raghavan D, Kronenberg H:Acute leukemia following chemotherapy includingetoposide for testicular carcinoma. AustN Z J Med 23:718-719, 1993.
42.
Cullen MH, Stenning SP, Parkinson MC:Short-course adjuvant chemotherapy in highriskstage I nonseminomatous germ cell tumorsof the testis. A Medical Research CouncilReport. J Clin Oncol 14:1106-1113, 1996.
43.
Gospodarowicz MK, Warde PR: Managementof stage I-II seminoma, in RaghavanD, Scher HI, Leibel SA, Lange P (eds): Principlesand Practice of Genitourinary Oncology,pp 683-696. Philadelphia, Lippincott-Raven,1997.
44.
Foster RS: Surgery of the retroperitoneum,in Raghavan D (ed): American CancerSociety Atlas of Germ Cell Tumors. Toronto,BC Decker, in press.
45.
Stoter G, Loehrer PJ Sr, Levi J: Managementof stage II and good risk stage III and IVnonseminomatous germ cell tumors, in RaghavanD, Scher HI, Leibel S, et al (eds): Principlesand Practice of Genitourinary Oncology,pp 715-721. Philadelphia, Lippincott-Raven,1997.
46.
Williams SD, Stablein DM, Einhorn LH,et al: Immediate adjuvant chemotherapy versusobservation with treatment at relapse inpathological stage II testicular cancer. N EnglJ Med 317:1433-1438, 1987. Horwich A, Mason M, Hendry W: Testiculartumours, in Souhami RL, Tannock I,Hohenberger P, et al: Oxford Textbook ofOncology, 2nd edition, pp 2002-2035. NewYork, Oxford University Press, 2002.
47.
Bosl GJ, Geller NL, Cirrincione C, et al:Multivariate analysis of prognostic variables inpatients with metastatic testicular cancer. CancerRes 43:3403-3407, 1983.
48.
Bajorin D, Katz A, Chan E, et al: Comparisonof criteria for assigning germ cell tumorpatients to “good-risk” and “poor-risk”studies. J Clin Oncol 6:786-792, 1986.
49.
Birch R, Williams S, Cone A, et al: Prognosticfactors for favorable outcome in disseminatedgerm cell tumors. J Clin Oncol4:400-407, 1988.
50.
Levi JA, Thomson D, Sandeman T, et al:A prospective study of cisplatin-based combinationchemotherapy in advanced germ cellmalignancy: Role of maintenance and longtermfollow-up. J Clin Oncol 6:1154-1160,1988.
51.
Mead GM, Stenning SP, Parkinson MC,et al: The Second Medical Research Councilstudy of prognostic factors in nonseminomatousgerm cell tumors. J Clin Oncol 10:85-94,1992.
52.
Levi JA, Thomson D, Sandeman T, et al:A prospective study of cisplatin-based combinationchemotherapy in advanced germ cellmalignancy: Role of maintenance and longtermfollow-up. J Clin Oncol 6:1154-1160,1988.
53.
Bosl GJ, Geller NL, Vogelzang NJ, et al:Alternating cycles of etoposide plus cisplatinand VAB-6 in the treatment of poor-risk patientswith germ cell tumors. J Clin Oncol 5:436-440, 1987.
54.
International Germ Cell Cancer CollaborativeGroup: International germ cell consensusclassification: A prognostic factor-basedstaging system for metastatic germ cell cancers.J Clin Oncol 15:594-603, 1997.
55.
Williams SD, Birch R, Einhorn LH, et al:Treatment of disseminated germ-cell tumorswith cisplatin, bleomycin, and either vinblastineor etoposide. N Engl J Med 316:1435-1440, 1987.
56.
Levi JA, Raghavan D, Harvey V, et al:The importance of bleomycin in combinationchemotherapy for good prognosis germ cellcarcinoma. Australasian Germ Cell Trial Group.J Clin Oncol 11:1300-1305, 1993.
57.
Loehrer PJ Sr, Johnson D, Elson P, et al:Importance of bleomycin in favorable-prognosisdisseminated germ cell tumors: An EasternCooperative Oncology Group Trial. J ClinOncol 13:470-476, 1995.
58.
deWit R, Stoter G, Kaye SB, et al:Importance of bleomycin in combination chemotherapyfor good-prognosis testicular nonseminoma:A randomized study of theEuropean Organization for Research andTreatment of Cancer Genitourinary Tract CancerCooperative Group. J Clin Oncol 15:1837-1843, 1997.
59.
Bajorin DF, Bosl GJ: Bleomycin in germcell tumor therapy: Not all regimens are createdequal. J Clin Oncol 15:1717-1719, 1997.
60.
Einhorn LH, Williams SD, Loehrer PJ,et al: Evaluation of optimal duration of chemotherapyin favorable prognosis disseminatedgerm cell tumors: A Southeastern CancerStudy Group protocol. J Clin Oncol 7:387-391, 1989.
61.
Horwich A, Dlejfer DT, Fossa SD, et al:Randomized trial of bleomycin, etoposide, andcisplatin compared with bleomycin, etoposide,and carboplatin in good-prognosis metastaticnonseminomatous germ cell cancer: A multiinstitutionalMedical Research Council/EuropeanOrganization for Research and Treatmentof Cancer Trial. J Clin Oncol 15:1844-1852,1997.
62.
Levine EG, Raghavan D: Treatment ofrefractory testis cancer: Salvage or savagechemotherapy? Eur J Cancer 27:932-936,1991.
63.
Nichols CR, Williams SD, Loehrer PJ,et al: Randomized study of cisplatin dose intensityin poor-risk germ cell tumors: A SoutheasternCancer Study Group and SouthwestOncology Group protocol. J Clin Oncol 9:1163-1172, 1991.
64.
Margolin K, Doroshow JH, Ahn C, et al:Treatment of germ cell cancer with two cyclesof high-dose ifosfamide, carboplatin, and etoposidewith autologous tem-cell support. J ClinOncol 14:2631-2637, 1996.
65.
Margolin K: High-dose chemotherapyand stem-cell support in the treatment of poorriskgerm cell cancer, in Raghavan D (ed):Atlas of Germ Cell Tumors. Toronto, BC Decker,in press.
66.
Motzer RJ, Mazumdar M, Bosl GJ, et al:High-dose carboplatin, etoposide and cyclophosphamidefor patients with refractory germcell tumors: Treatment results and prognosticfactors for survival and toxicity. J Clin Oncol 14:1098-1105, 1996.
67.
Motzer RJ, Mazumdar M, Bajorin DF, etal: High-dose carboplatin, etoposide, and cyclophosphamidewith autologous bone marrowtransplantation in first-line therapy forpatients with poor-risk germ cell tumors. J ClinOncol 15:2546-2552, 1997.
68.
Newlands ES, Begent RH, Rustin GJ,et al: Further advances in the management ofmalignant teratomas of the testis and other sites.Lancet 1:948-951, 1983.
69.
Bower M, Newlands ES, Holden L, et al:Treatment of men with metastatic non-seminomatousgerm cell tumours with cyclicalPOMB/ACE chemotherapy. Ann Oncol 8:477-483, 1997.
70.
Motzer RJ, Bajorin DF, Schwartz LH,et al: Phase II trial of paclitaxel shows antitumoractivity in patients with previously treatedgerm cell tumors. J Clin Oncol 12:2277-2283, 1994.
71.
Bokemeyer C, Gerl A, Schoffski P, et al:Gemcitabine in patients with relapsed or cisplatin-refractory testticular cancer. J Clin Oncol17:512-516, 1999.
72.
Einhorn LH, Stender MJ, Williams SD:Phase II trial of gemcitabine in refractory germcell tumors. J Clin Oncol 17:509-511, 1999.
73.
Motzer RJ, Sheinfeld J, Mazumdar M, etal: Paclitaxel, ifosfamide, and cisplatin secondlinetherapy for patients with relapsed testiculargerm cell cancer. J Clin Oncol18:2413-2418, 2000.
74.
Hinton S, Catalano P, Einhorn LH, et al:Phase II study of paclitaxel plus gemcitabine inrefractory germ cell tumors (E9897): A trial ofthe Eastern Cooperative Oncology Group.J Clin Oncol 20:1859-1863, 2002.
75.
Rick O, Bokemeyer C, Beyer J, et al:Salvage treatment with paclitaxel, ifosfamide,and cisplatin plus high-dose carboplatin, etopoisde,and thiotepa followed by autologousstem-cell rescue in patients with relapsed orrefractory germ cell cancer. J Clin Oncol 19:81-88, 2001.
76.
Boyer M, Raghavan D: Toxicity of treatmentof germ cell tumors. Semin Oncol 19:128-142, 1992.
77.
Boyer MJ, Roth BJ: Toxicity of treatmentof germ cell tumors, in Raghavan D,Scher HI, Leibel S, et al (eds): Principles andPractice of Genitourinary Oncology, pp 754-764. Philadelphia, Lippincott-Raven, 1997.
78.
van Leeuwen FE, Stiggelbout AM, vanden Belt-Dusebout A, et al: Second cancer riskfollowing testicular cancer: A follow-up studyof 1909 patients. J Clin Oncol 11:415-424,1993.
79.
Travis LB, Curtis RE, Storm H, et al:Risk of second malignant neoplasms amonglong-term survivors of testicular cancer. J NatlCancer Inst 89:1429-1439, 1997.
80.
Meinardi MT, Gietema JA, van der GraafWT, et al: Cardiovascular morbidity in longtermsurvivors of metastatic testicular cancer.J Clin Oncol 18:1725-1732, 2000.













