Epigenetics in Cancer: What's the Future?
The understanding that epigenetic changes are prevalent in cancer and play a causative role in its biology has led to the development of new therapeutic approaches that target the epigenetic machinery.
Epigenetics is a rapidly expanding field that focuses on stable changes in gene expression that are not accompanied by changes in DNA sequence and that are mediated primarily by DNA methylation and histone modifications. Disruption of the epigenome is a fundamental mechanism in cancer, and several epigenetic drugs that have proved to prolong survival and to be less toxic than conventional chemotherapy were recently approved by the FDA for cancer treatment. These include azacitidine (Vidaza), decitabine (Dacogen), vorinostat (Zolinza), and romidepsin (Istodax). Promising results of combination clinical trials with DNA methylation inhibitors and histone deacetylase inhibitors have recently been reported, and data are emerging that describe molecular determinants of clinical responses. Despite significant advances, challenges remain, including a lack of predictive markers, unclear mechanisms of response and resistance, and rare responses in solid tumors. Preclinical studies are ongoing with novel classes of agents that target various components of the epigenetic machinery. In this review, we focus on recent clinical and translational data in the epigenetics field that have potential in cancer therapy.
Epigenetics is defined as the study of stable changes in gene expression that are not accompanied by changes in DNA sequence.[1] Epigenetic changes are important biological processes with relevance to all multicellular organisms. Studies in various models have shown that epigenetic regulation is critical for proper embryogenesis and development. Several mechanisms of epigenetic change have been described, and all seem to be interdependent to some degree. DNA methylation, posttranslational modifications of histones, and chromatin remodeling enzymes mediate epigenetic changes in many organisms. It has become clear that disruption of the epigenetic machinery plays a fundamental role in cancer development. Tumors often exhibit global hypomethylation, hypermethylation of CpG islands, and genome-wide alterations in the levels of histone modifications. These abnormalities are associated with widespread changes in gene expression, which are thought to contribute to tumor formation by affecting oncogenes and tumor suppressor genes.[1]
What Is Epigenetic Therapy?
The understanding that epigenetic changes are prevalent in cancer and play a causative role in its biology has led to the development of new therapeutic approaches that target the epigenetic machinery. The first successful drugs developed as epigenetic agents were DNA methyltransferase inhibitors; these were followed by histone deacetylase inhibitors (HDIs). Both classes of drugs aim at reversing gene silencing and demonstrate antitumor activity in vitro and in vivo. Several other classes of drugs have been developed that target various other components of the epigenetic machinery; one such class is the histone methyltransferases, with new drugs in this class currently in early preclinical development (Table 1).
TABLE 1


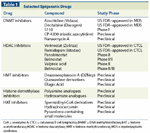
Selected Epigenetic Drugs
What Has Been Done?
The inhibitors of DNA methylation used clinically are nucleoside analogues that get converted into deoxy-nucleotide-triphosphates (dNTPs) and become incorporated into DNA in place of cytosine during DNA replication. They trap all DNA methyltransferases and target them for degradation. At low doses these drugs do not inhibit proliferation; they reactivate gene expression and have shown clinical activity as anticancer agents. Azacitidine (AZA; Vidaza) was the first hypomethylating agent approved by the FDA; its approval, in 2004, for the treatment of myelodysplastic disorders and leukemia, was followed by the approval, in 2006, of decitabine (DAC; 5-aza-2′-deoxyctidine; Dacogen).[2,3] Both drugs produce remissions or clinical improvements in more than 30% of patients treated. Features of responses have included the requirement for multiple cycles of therapy, slow response, and relatively few side effects. On the molecular level, demethylation, gene reactivation, and clonal elimination were observed in treated patients.[4] The data in myelodysplastic syndrome (MDS) represent a proof-of-principle for epigenetic therapy for cancer, in particular in myeloid disorders.
HDIs work by preventing histone deacetylation, thereby facilitating an open chromatin structure and leading to the activation of genes, including the p21 cyclin-dependent kinase inhibitor. Several HDIs have shown antitumor activity in vitro with little toxicity in preclinical studies, suggesting selectivity for neoplastic cells. This has prompted the development of additional compounds, many of which have entered phase 1 trials in various malignancies. Two HDIs have shown particular efficacy against cutaneous T cell lymphoma (CTCL)-response rates of over 30%-which led to the FDA approval of vorinostat (suberoylanilide hydroxamic acid; Zolinza) in 2007 and of romidepsin (depsipeptide; Istodax) in 2009.[5,6]
TABLE 2


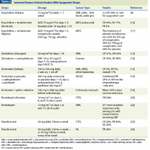
Selected Epigenetic Drugs
Recent Clinical Trials
So far, all four FDA-approved epigenetic drugs have shown the greatest efficacy in hematopoietic malignancies. There is no known reason why this should be true, or why solid tumors would not respond as well. Older studies suggested a lack of activity for these agents in various solid tumors; however, most testing was done at high doses, with short exposures, and in patients with refractory disease-conditions also associated with lack of response in hematologic malignancies. There may be pharmacologic or pharmacodynamic reasons that favor responses in leukemias (eg, drug uptake, proportion of proliferating cells), but the activity of these agents in solid tumors certainly deserves a second look. Here, we describe recent clinical advances with FDA-approved and investigational epigenetic agents, both in hematologic malignancies and in solid tumors (Table 2).
AZA
AZA was recently tested in a phase III trial in MDS. The overall response rate in the first report was 21%, with documented improved survival compared with either supportive care or chemotherapy.[7,8] In a subset analysis, improved survival was also seen in patients with acute myeloid leukemia (AML).[9] The combination of AZA with lenalidomide (Revlimid) in high-risk MDS showed a promising 67% overall response rate in a phase I study.[10] A phase II study demonstrated a decrease in platelet transfusion dependence for patients receiving AZA plus romiplostim (Nplate).[11]
DAC
Several phase II trials have demonstrated substantial activity for DAC as a single agent in AML, resulting in a complete response (CR) in 24% to 52% of patients; the drug appears to be especially promising for older patients.[12,13] A recent early combination trial of DAC with carboplatinum showed some activity in platinum-resistant ovarian cancer and demonstrated demethylating activity, thus justifying further clinical efficacy testing.[14]
Vorinostat
Results of a phase I study of vorinostat in patients with leukemias have been reported.[15] Seven patients (17%) achieved a CR, a CR with incomplete blood count recovery, or hematological improvement; all had AML. Promising results were recently shown in advanced non–small-cell lung cancer (NSCLC), where vorinostat vs placebo was used in addition to the combination of carboplatinum and paclitaxel.[16] Vorinostat significantly enhanced the efficacy of chemotherapy in patients with advanced NSCLC (overall response rate, 34% vs 12.5%), and there was a trend toward improvement in median progression-free survival and overall survival in the vorinostat arm. Modest responses were recently demonstrated in non-Hodgkin lymphoma and glioblastoma multiforme with vorinostat used as a single agent; it would be interesting to test it in combination regimens in the future.[17,18] A phase I study in patients with colorectal and gastric carcinomas tested the first clinical combination of an HDI with therapeutic radiation, and it found the combination to be well tolerated and safe. Change in tumor volume was evaluable in 14 patients and showed considerable variation, ranging from a 54% reduction to a 28% increase (mean, 26% reduction). These data will underpin future chemoradiation studies of vorinostat combined with radiotherapy.[19]
Romidepsin
Final results of a phase II multicenter study in CTCL have been published; a 34% response rate was achieved in a refractory setting, making the use of romidepsin an important therapeutic option for this disease.[20] Romidepsin showed minimal activity in MDS[21] and low-level activity in AML in a phase I trial; interestingly, responses were seen only in a subgroup of core binding factor leukemias.[21]
Panobinostat
Panobinostat is one of the most potent HDIs in vitro, and preliminary studies have shown activity in CTCL.[23] More recently, promising phase I results were demonstrated in refractory Hodgkin lymphoma [24] and in prostate cancer; a decline in PSA levels was reported in 60% of patients in an early trial of panobinostat in combination with docetaxel (Taxotere).[25]
Belinostat
An early phase I study of belinostat, another potent HDI, in combination with carboplatinum and/or paclitaxel was recently conducted; this trial showed some activity of belinostat in various solid tumors, although it is unclear whether the addition of belinostat was superior to chemotherapy alone.[26] Another phase II study showed some activity in ovarian cancer.[27]
Valproic acid
Valproic acid is a short-chain fatty acid that is a weak HDI. A single-agent study demonstrated minor responses in patients with MDS.[28] Most of the clinical data on valproic acid have come from combination studies. A larger trial of valproic acid and epirubicin/5-FU/cyclophosphamide showed 22% partial responses in solid tumors.[29] Another phase I/II trial of valproic acid with a topoisomerase I inhibitor in metastatic melanoma showed 47% disease stabilization.[30]
Problems, Mechanisms and Potential Solutions
Epigenetic therapy presents several clinical and translational challenges. In particular, while DNA methylation inhibitors and HDIs are considered targeted agents, this approach is generally nonspecific because of the numerous downstream effects on gene expression. Also, despite documented epigenetic effects, there is no consensus as to whether or not the responses are mediated strictly via epigenetic mechanisms. Thus, translational research will be critical to clarify the precise mechanisms of the in vivo action of the drugs-and mechanisms of resistance. Furthermore, as new data emerge regarding the response markers and mechanisms of action of these drugs, combination therapy involving epigenetic agents together with conventional or targeted agents is increasingly seen as an attractive opportunity. Therefore, preclinical combinatorial resistance or synthetic lethality screens are likely to be instrumental in selecting the most potent drugs and combinations.
While several studies have documented the epigenetic effects of decitabine and azacitidine in vitro and in vivo,[31-33] other studies have elucidated other mechanisms. As an example, it was recently shown that DAC upregulates p21 and induces cell cycle arrest in a DNA methyltransferase (DNMT)-independent fashion through DNA damage ATM/P53 axis, implicating the role of these pathways in responses.[34] However, the same group has demonstrated that neither DNA damage nor methylation changes were predictive of response to demethylating therapy.[35] Further, decitabine effects were studied by whole-genome expression assays in melanoma cells; p21 induction was observed and WNT pathway activation was found to be contributing to resistance. Interestingly, inhibition of the proteasome increased the growth arrest of DAC-treated cells, implying that proteasome inhibitors might act in synergy with epigenetic modifiers.[36] An interesting study looking at cytotoxic CD8+ T-cell responses to MAGE antigens has demonstrated that epigenetic therapy with a DNMT inhibitor induced such responses in vivo and was correlating with clinical responses in 8 out of 11 patients; thus, it is possible that unmasking MAGE antigens with epigenetic drugs caused immune clearance of leukemic cells.[37] Epigenetic drugs are known to induce differentiation, and a study recently described how both cytarabine and DAC were able to induce differentiation of embryonic cancer stem cells via caspase-mediated degradation of EZH2 as well as NANOG and OCT-4 stem cell factors, suggesting yet another mechanism of action of nucleoside analogues and epigenetic drugs operating at the stem cell level.[38]
To resolve these issues regarding the mechanism(s) of action of epigenetic drugs, the identification of predictors or markers of clinical responses is of paramount importance. However, we are at an early stage in understanding the process of identifying predictors/response markers. A recent study of elderly patients with AML who were treated with decitabine showed that higher pretreatment levels of microRNA-29b (miR-29b), previously shown to target DNMTs, correlated with clinical responses; thus, if these results are validated, miR-29b levels could be used for stratification of patients.[12] Several HDI studies were recently published that proposed explanations of responses seen in CTCLs. Using a genome-wide loss of function screen with a short hairpin RNA (shRNA) library, HR23B, a component of the proteasome, was identified as a determinant of HDI sensitivity.[39] HR23B is expressed at high levels in CTCL, and there was a correlation between HR23B expression and clinical response to HDIs.[40] Further, a mechanistic study of panobinostat showed that this agent induces CXCR4 degradation via the proteasomal pathway and has synergy with CXCR4 inhibition, supporting the rationale for testing such a combination in vivo.[41] The same research group reported another combination study of the EZH2 inhibitor 3-deazaneplanocin A (DZNep) with panobinostat. This study demonstrated induction of more apoptosis in AML cells with the combination therapy than with either agent alone; the combination also prolonged survival in a mouse model.[42] Another study of neuroblastoma cells exhibiting silencing of caspase-8 demonstrated that the combination of various HDIs with interferon-gamma was effective in overcoming the resistance of such cells to tumor necrosis factor–related apoptosis-inducing ligand (TRAIL), thus prompting further investigations.[43] Finally, a study addressing fundamental mechanisms of drug resistance identified a subpopulation of cancer cells that upregulated insulin-like growth factor 1(IGF-1) signaling and acquired an altered chromatin state via RBP2/KDM5A/Jarid1A demethylase upregulation and concomitant decreases in H3K4 methylation and H3K14 acetylation. Interestingly, drug-tolerant cells could be ablated by IGF-1 or HDIs.[44] The role of RBP2 in mediating clinical sensitivity to epigenetic drugs needs testing; this approach may yield a novel therapeutic opportunity to use epigenetic therapy to reverse drug resistance in patients in whom resistance to chemotherapy is developing.
Novel Epigenetic Drugs
There is great enthusiasm in both academia and industry for developing new epigenetic compounds. Some of the recent preclinical data regarding several of these potential drugs are summarized below.
DNMT inhibitors
S110 is a 5-aza-2'-deoxycytidine–containing dinucleotide that releases decitabine intracellularly and has shown improved stability in vitro, as well as demonstrating hypomethylating and anti-tumor effects in a cellular model.[45] Clinical trials with S110 are planned for the near future. CP-4200, an elaidic acid derivative of azacitidine was recently developed. This compound has demonstrated strong epigenetic effects in cell lines; it causes depletion of DNA methyltransferase, genome-wide DNA demethylation, and robust reactivation of epigenetically silenced tumor suppressor genes.[46] Importantly, the cellular uptake of CP-4200 is substantially less dependent on the nucleoside transporters, and the compound showed significantly higher antitumoral activity than azacitidine in an orthotopic mouse tumor model of acute lymphocytic leukemia. Thus, these early data suggest that elaidic acid modification may improve the therapeutic efficacy of azacitidine. The first DNMT3B-selective inhibitor, nanaomycin A, has been described; this compound exhibited genomic demethylation and antiproliferative effects in different cell lines.[47]
HDIs
Several new HDIs have recently shown promise in preclinical testing. Resminostat is a potent inhibitor of HDACs 1, 3 and 6, and in low micromolar concentrations it abrogated cell growth and strongly induced apoptosis in multiple myeloma cell lines and primary cells. It has shown synergistic effects in vitro in combinations with melphalan and proteasome inhibitors.[48] AR-42 is a novel HDI that recently showed efficacy against B cells in vivo and in vitro. It demonstrated inhibition of primary leukemia and lymphoma cell lines, and it significantly reduced leukocyte counts and/or prolonged survival in three separate mouse models of B-cell malignancy, without evidence of toxicity.[49] Novel HDIs have also been shown to be active against solid tumors, in particular lung cancer. For instance, the novel pan-HDAC inhibitor, OSU-HDAC-44, exhibits three to four times more effectiveness than vorinostat in suppressing cell viability in various NSCLC cell lines. It inhibited cytokines, induced mitochondria-mediated apoptosis, and inhibited xenograft tumor growth; based on these data, it is a promising chemotherapeutic drug for NSCLC.[50]
Histone methyltransferase inhibitors
EZH2 is a histone methyltransferase that is overexpressed in multiple cancer types and that has an activating mutation in lymphomas that makes it an attractive therapeutic target.[51,52] DZNep has been shown to deplete components of the PRC2 complex, including EZH2, and to induce apoptosis in cancer cells.[53] Inhibition of EZH2 in glioblastoma stem cells by DZNep resulted in impairment of cell renewal and of the tumor-initiating capacity of these cells, in part via c-myc downregulation.[54] DZNep has also been shown to be especially effective in killing BRCA-deficient breast cancer cells.[55] Another histone methyltransferase, G9a, which targets a different residue, has previously been shown to be important for cancer cell survival.[56] Two inhibitors of G9a, the diazepin-quinazolin-amine derivative BIX-01294 and the 2,4-diamino-6,7-dimethoxyquinazoline derivative named UNC0224, were recently described but have not yet been tested in vivo.[57-59] Coactivator-associated arginine methyltransferase 1 (CARM1) is an arginine methyltransferase that has been implicated in prostate cancer development.[60] Ellagic acid has recently been shown to be a specific CARM1 inhibitor that reduced H3R17 levels.[61] It is expected that several histone methyltransferase inhibitors will enter clinical trials in the next few years.
Histone demethylase inhibitors
To date, two classes of histone lysine demethylases have been identified. One class includes the lysine-specific demethylases LSD1 and LSD2, and a second comprises the recently discovered Jumonji domain–containing protein (JMJD) histone demethylases. LSD1 has been shown to be overexpressed and/or critical for cell proliferation in prostate cancer,[62-64] breast cancer,[65] and neuroblastoma.[66] LSD1 inhibitors were recently developed and have demonstrated reactivation of silenced genes as well as inhibition of colon cancer growth.[67-69] Similarly, JMJD demethylases have been implicated in cell growth in prostate,[63] breast,[70] and esophageal cancers.[71] JMJD inhibitors have also recently been described; these have demonstrated inhibition of prostate and colon cancer cell growth.[72] Further characterization of these compounds is needed in anticipation of potential clinical trials.
REFERENCE GUIDE
Therapeutic Agents
Mentioned in This Article
5-FU
AR-42
Azacitidine (Vidaza)
Belinostat
BIX-01294
C646
Carboplatinum
CP-4200
Cyclophosphamide
Cytarabine
Decitabine (Dacogen)
Docetaxel (Taxotere)
DZNep
Ellagic acid
Epirubicin
Hydrazinocurcumin
Interferon-gamma
Lenalidomide (Revlimid)
Nanaomycin A
OSU-HDAC-44
Paclitaxel
Panobinostat
Resminostat
Romidepsin (Istodax)
Romiplostim (Nplate)
S110
UNC0224
Valproic acid
Vorinostat (Zolinza)
Brand names are listed in parentheses only if a drug is not available generically and is marketed as no more than two trademarked or registered products. More familiar alternative generic designations may also be included parenthetically.
Histone acetyltransferase (HAT) inhibitors
HATs are involved in many critical cellular proliferation pathways, and several chromosomal translocations observed in leukemias directly disrupt these enzymes. Translocation t(8;16)(p11;p13), a cytogenetic hallmark for the M4/M5 subtype of AML, fuses the CREB-binding protein (CBP) on chromosome 16 with a chromosome 8 HAT, MOZ. MOZ-CBP might induce leukemia by antagonizing the function of the AML1 complex,[73] since MOZ-driven acetylation plays a role in controlling a desirable balance between proliferation and differentiation during hematopoiesis.[74] Further, t(11;16)(q23;p13.3), a common translocation in acute leukemia and secondary myelodysplasia, results in a fusion of the MLL gene with the CBP gene. The chimeric fusion product retains the HAT domain of CBP and may lead to leukemia by promoting histone acetylation of genomic regions targeted by MLL, which in turn leads to transcriptional deregulation via aberrant chromatin organization.[75] These findings make HATs an attractive target for cancer therapy, and various inhibitors have been described, although few have been tested against cancer cells.[76,77] A recent study described a novel HAT inhibitor, a derivative of spermidinyl–coenzyme A, which was shown to be effective as a cancer-specific chemo- and radiosensitizer.[78] Another study described the successful blocking of hyperacetylation of histone H3 in oral squamous cell cancers by a curcumin derivative, resulting in inhibited cancer cell growth.[79] Another HAT inhibitor, C646, was shown to inhibit lung and melanoma cancer cells without affecting a normal fibroblast cell line.[80]
Conclusions
The use of epigenetic drugs allows restoration of the expression of genes that are deregulated in cancer. These agents help patients with MDS live longer with fewer side effects than is possible with standard chemotherapy. Several early trials have shown promising results in patients with solid tumors, especially NSCLC, where vorinostat appears to enhance the efficacy of chemotherapy. HDIs are also now being tested as radiosensitizing agents. Components of the proteasome have been identified as likely sensitivity determinants for HDIs, whereas immune responses and degradation of stem cell factors have been reported to be likely sensitivity determinants for DNA methylation inhibitors. Finally, several novel epigenetic drugs targeting histone-modifying enzymes are emerging and have entered preclinical testing; hopes are high that these agents will be seen in clinical trials in the near future.
Financial Disclosure:Dr. Issa receives research support from Eisai, Merck, and Celgene; and honoraria from Celgene, Novartis, and Johnson & Johnson. He serves as a consultant for GlaxoSmithKline and Syndax. Dr. Boumber has no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
References
1. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683-92.
2. Issa JP, Kantarjian HM, Kirkpatrick P: Azacitidine. Nat Rev Drug Discov. 2005;4:275-6.
3. Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794-1803.
4. Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res. 2007;13:1634-7.
5. Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Investig Drugs. 2007;16:1111-20.
6. Grant C, Rahman F, Piekarz R, et al. A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther. 2010;10:997-1008.
7. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223-32.
8. Fenaux P, Gattermann N, Seymour JF, et al. Prolonged survival with improved tolerability in higher-risk myelodysplastic syndromes: azacitidine compared with low dose ara-C. Br J Haematol. 2010;149:244-9.
9. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28:562-9.
10. Sekeres MA, List AF, Cuthbertson D, et al. Phase I combination trial of lenalidomide and azacitidine in patients with higher-risk myelodysplastic syndromes. J Clin Oncol. 2010;28:2253-8.
11. Kantarjian HM, Giles FJ, Greenberg PL, et al. Phase II study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood. 2010;116:3163-70.
12. Blum W, Garzon R, Klisovic RB, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci U S A. 2010;107:7473-8.
13. Cashen AF, Schiller GJ, O'Donnell MR, et al. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28:556-61.
14. Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043-53.
15. Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060-6.
16. Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:56-62.
17. Watanabe T, Kato H, Kobayashi Y, et al. Potential efficacy of the oral histone deacetylase inhibitor vorinostat in a phase I trial in follicular and mantle cell lymphoma. Cancer Sci. 2010;101:196-200.
18. Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group study. J Clin Oncol. 2009;27:2052-8.
19. Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459-64.
20. Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485-91.
21. Klimek VM, Fircanis S, Maslak P, et al. Tolerability, pharmacodynamics, and pharmacokinetics studies of depsipeptide (romidepsin) in patients with acute myelogenous leukemia or advanced myelodysplastic syndromes. Clin Cancer Res. 2008;14:826-32.
22. Odenike OM, Alkan S, Sher D, et al. Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clin Cancer Res. 2008;14:7095-101.
23. Ellis L, Pan Y, Smyth GK, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res. 2008;14:4500-510.
24. Dickinson M, Ritchie D, DeAngelo DJ, et al. Preliminary evidence of disease response to the pan deacetylase inhibitor panobinostat (LBH589) in refractory Hodgkin lymphoma. Br J Haematol. 2009;147:
97-101.
25. Rathkopf D, Wong BY, Ross RW, et al. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2010;66:181-9.
26. Lassen U, Molife LR, Sorensen M, et al. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br J Cancer. 2010;103:12-17.
27. Mackay HJ, Hirte H, Colgan T, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer. 2010;46:1573-9.
28. Kuendgen A, Strupp C, Aivado M, et al. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004;104:1266-9.
29. Munster P, Marchion D, Bicaku E, et al. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res. 2009;15:2488-96.
30. Daud AI, Dawson J, Deconti RC, et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res. 2009;15:2479-87.
31. Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52-7.
32. Yang AS, Doshi KD, Choi SW, et al. DNA methylation changes after 5-aza-2'-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66:5495-503.
33. Mund C, Hackanson B, Stresemann C, et al. Characterization of DNA demethylation effects induced by 5-aza-2'-deoxycytidine in patients with myelodysplastic syndrome. Cancer Res. 2005;65:7086-90.
34. Jiemjit A, Fandy TE, Carraway H, et al. p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage. Oncogene. 2008;27:3615-23.
35. Fandy TE, Herman JG, Kerns P, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114:2764-73.
36. Halaban R, Krauthammer M, Pelizzola M, et al. Integrative analysis of epigenetic modulation in melanoma cell response to decitabine: clinical implications. PLoS One. 2009;4:e4563.
37. Goodyear O, Agathanggelou A, Novitzky-Basso I, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116:1908-18.
38. Musch T, Oz Y, Lyko F, Breiling A. Nucleoside drugs induce cellular differentiation by caspase-dependent degradation of stem cell factors. PLoS One. 2010;5:e10726.
39. Fotheringham S, Epping MT, Stimson L, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15:57-66.
40. Khan O, Fotheringham S, Wood V, et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci U S A. 2010;107:6532-7.
41. Mandawat A, Fiskus W, Buckley KM, et al. Pan-histone deacetylase (HDAC) inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic anti-myeloid activity in combination with CXCR4 antagonists. Blood. 2010;Epub ahead of print.
42. Fiskus W, Wang Y, Sreekumar A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733-43.
43. Hacker S, Dittrich A, Mohr A, et al. Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene. 2009;28:3097-110.
44. Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80.
45. Yoo CB, Jeong S, Egger G, et al. Delivery of 5-aza-2'-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400-408.
46. Brueckner B, Rius M, Markelova MR, et al. Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol Cancer Ther. 2010;9:1256-64.
47. Kuck D, Caulfield T, Lyko F, Medina-Franco JL. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther. 2010;9:3015-23.
48. Mandl-Weber S, Meinel FG, Jankowsky R, et al. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol. 2010;149:518-28.
49. Lucas DM, Alinari L, West DA, et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS One. 2010;5:e10941.
50. Tang YA, Wen WL, Chang JW, et al. A novel histone deacetylase inhibitor exhibits antitumor activity via apoptosis induction, F-actin disruption and gene acetylation in lung cancer. PLoS One. 2010;5:e12417.
51. Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-9.
52. Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181-5.
53. Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050-63.
54. Suva ML, Riggi N, Janiszewska M, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69:9211-8.
55. Puppe J, Drost R, Liu X, et al. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb repressive complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009;11:R63.
56. Kondo Y, Shen L, Ahmed S, et al. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One. 2008;3:e2037.
57. Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950-3.
58. Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:
5844-57.
59. Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473-81. 60. Majumder S, Liu Y, Ford OH, III, et al. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate. 2006;66:1292-1301.
61. Selvi BR, Batta K, Kishore AH, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17. J Biol Chem. 2010;285:7143-52.
62. Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436-9.
63. Wissmann M, Yin N, Muller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347-53.
64. Kahl P, Gullotti L, Heukamp LC, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341-7.
65. Lim S, Janzer A, Becker A, et al. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31:512-20.
66. Schulte JH, Lim S, Schramm A, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065-71.
67. Huang Y, Stewart TM, Wu Y, et al. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217-28.
68. Huang Y, Greene E, Murray ST, et al. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007;104:8023-8.
69. Huang Y, Marton LJ, Woster PM, Casero RA. Polyamine analogues targeting epigenetic gene regulation. Essays Biochem. 2009;46:95-110.
70. Liu G, Bollig-Fischer A, Kreike B, et al. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28:4491-500.
71. Cloos PA, Christensen J, Agger K, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307-11.
72. Hamada S, Suzuki T, Mino K, et al. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629-38.
73. Kitabayashi I, Aikawa Y, Nguyen LA, et al. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. EMBO J. 2001;20:7184-96.
74. Perez-Campo FM, Borrow J, Kouskoff V, Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood. 2009;113:4866-74.
75. Sobulo OM, Borrow J, Tomek R, et al. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc Natl Acad Sci U S A. 1997;94:8732-7.
76. Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23-36.
77. Vernarecci S, Tosi F, Filetici P. Tuning acetylated chromatin with HAT inhibitors: a novel tool for therapy. Epigenetics. 2010;5:105-11.
78. Bandyopadhyay K, Baneres JL, Martin A, et al. Spermidinyl-CoA-based HAT inhibitors block DNA repair and provide cancer-specific chemo- and radiosensitization. Cell Cycle. 2009;8:2779-88.
79. Arif M, Vedamurthy BM, Choudhari R, et al. Nitric oxide-mediated histone hyperacetylation in oral cancer: target for a water-soluble HAT inhibitor, CTK7A. Chem Biol. 2010;17:903-13.
80. Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471-82.














