Hereditary vs Familial Pancreatic Cancer: Associated Genetic Syndromes and Clinical Perspective
Pancreatic ductal adenocarcinoma is a disease marked by high rates of mortality, with only about 7% of patients surviving 5 years after diagnosis. Here, the authors present a demonstrative case and review the available data on hereditary and familial PDAC.


Copur is a medical oncologist/hematologist at Morrison Cancer Center, Mary Lanning Healthcare in Hastings, Nebraska, and is a professor at the University of Nebraska Medical Center. He is also editor-at-large and a Community Oncology Advisory Board member at ONCOLOGY®.



Talmon is a professor in the Department of Pathology and Microbiology at the University of Nebraska Medical Center, specializing in gastrointestinal, renal, and placental pathology.



Wedel is a staff pathologist at Mary Lanning Healthcare.



Hart is a radiologist at Mary Lanning Healthcare.



Merani is assistant professor, transplantation surgery, University of Nebraska.


Vargas is assistant professor, transplantation surgery, University of Nebraska.

ABSTRACT: Pancreatic ductal adenocarcinoma (PDAC) is a disease marked by high rates of mortality; it is mostly incurable at the time of diagnosis. Only about 7% of patients survive 5 years after diagnosis. Diagnosis at a late stage and rapid progression with minimal response to available treatments are the main reasons for this poor outcome. It is crucial to identify individuals at high risk of developing PDAC so preventive and early detection measures can be employed. Approximately 10% to 15% of PDAC cases have a hereditary or familial basis. In the majority of PDAC cases, no main causative gene has been identified, but several known germline pathogenic mutations have been shown to be related to an increased risk of this cancer. The presence of 2 or more patients with pancreatic cancer within the circle of first-degree relatives, without the presence of a causative germline mutation, is defined as familial pancreatic cancer; this accounts for 4% to 10% of PDAC. Based on the growing evidence supporting the benefit of germline genetic testing in patients with PDAC, both the American Society of Clinical Oncology and the National Comprehensive Cancer Network recently updated their guidelines to include recommendations around genetic testing for patients with pancreatic cancer. However, there is no general consensus on the group of patients and individuals who should be studied and screened. We present a demonstrative case and review the available data on hereditary and familial PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has one of the worst prognoses among major cancers. In 2020 in the United States, it is expected to be the third-leading cause of cancer mortality, following lung and colorectal cancer, with 47,050 deaths in both genders combined (Table 1).1


Table 1. Estimated Deaths by Cancer Type, Both Sexes Combined. American Cancer Society, 2020
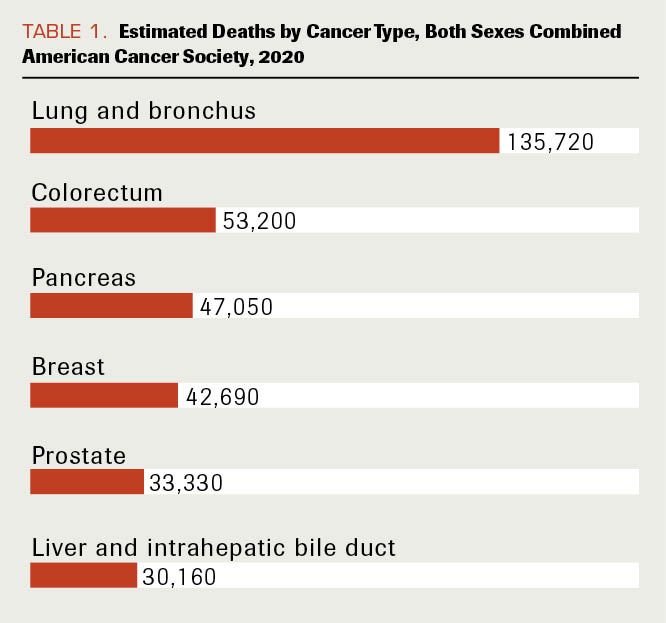
The average lifetime risk of pancreatic cancer is about 1 in 65 (1.5%), with a 5-year survival rate of 7%.2 Although incidence is roughly equal in both genders, African Americans have a higher incidence than any other racial group.3 Older age is among the main risk factors for the development of PDAC; median age at diagnosis is 71 years.4 Apart from family history, which can influence the likelihood of PDAC development, other risk factors include tobacco and alcohol abuse, chronic pancreatitis, dietary factors, obesity, and type 2 diabetes mellitus.5-8 The incidence of PDAC in the United States has increased in recent years, possibly due to the aging population and the increasing prevalence of obesity.


Table 2. Hereditary/Familial Syndromes Associated With Pancreatic Ductal Adenocarcinoma
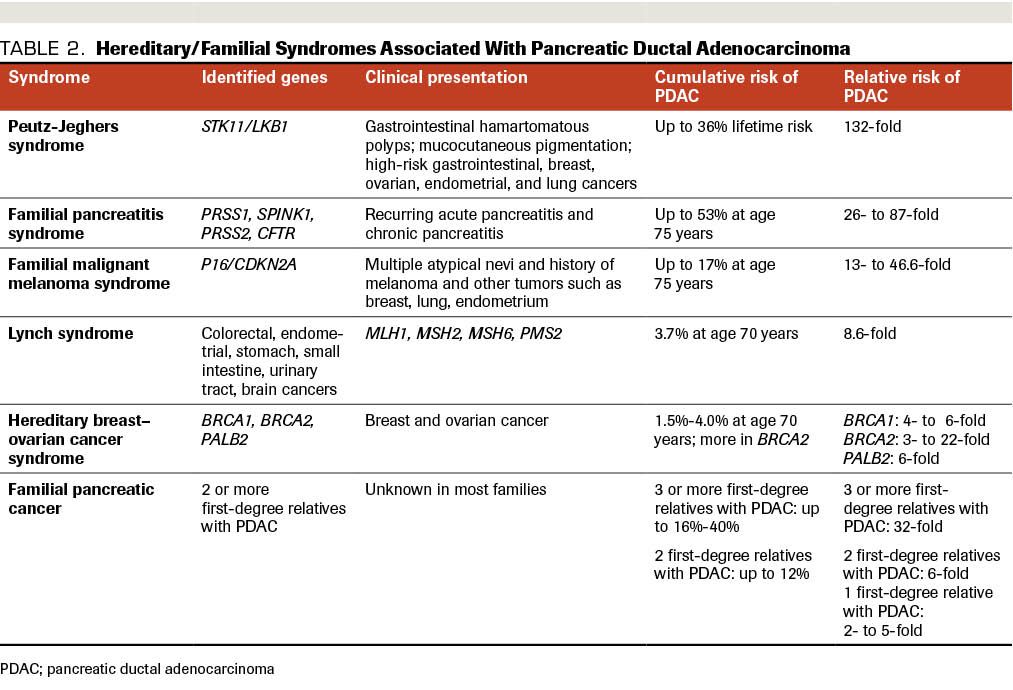
First-degree relatives of patients with PDAC have at least a 2-fold increased risk of developing the disease. The risk increases proportionally to the number of affected first-degree relatives.9-11 In nearly 10% to 15 % of all PDAC cases, a hereditary cancer predisposition syndrome can be implicated. Among the genetic syndromes associated with PDAC are Peutz-Jeghers,12 hereditary pancreatitis,13-16 familial atypical multiple mole melanoma,17,18 hereditary breast–ovarian cancer,19-22 and hereditary nonpolyposis colorectal cancer (Lynch) syndromes23,24 (Table 2). However, in the remaining 85% to 90% of cases with familial aggregation of PDAC, there is a lack of such a defined hereditary cancer predisposition component.25 The presence of 2 or more patients with pancreatic cancer within the circle of first-degree relatives without an association to a known hereditary cancer genetic syndrome has been defined as familial pancreatic cancer, which accounts for 4% to 10% of PDAC. In the majority of families with PDAC, a responsible gene mutation cannot be identified.26


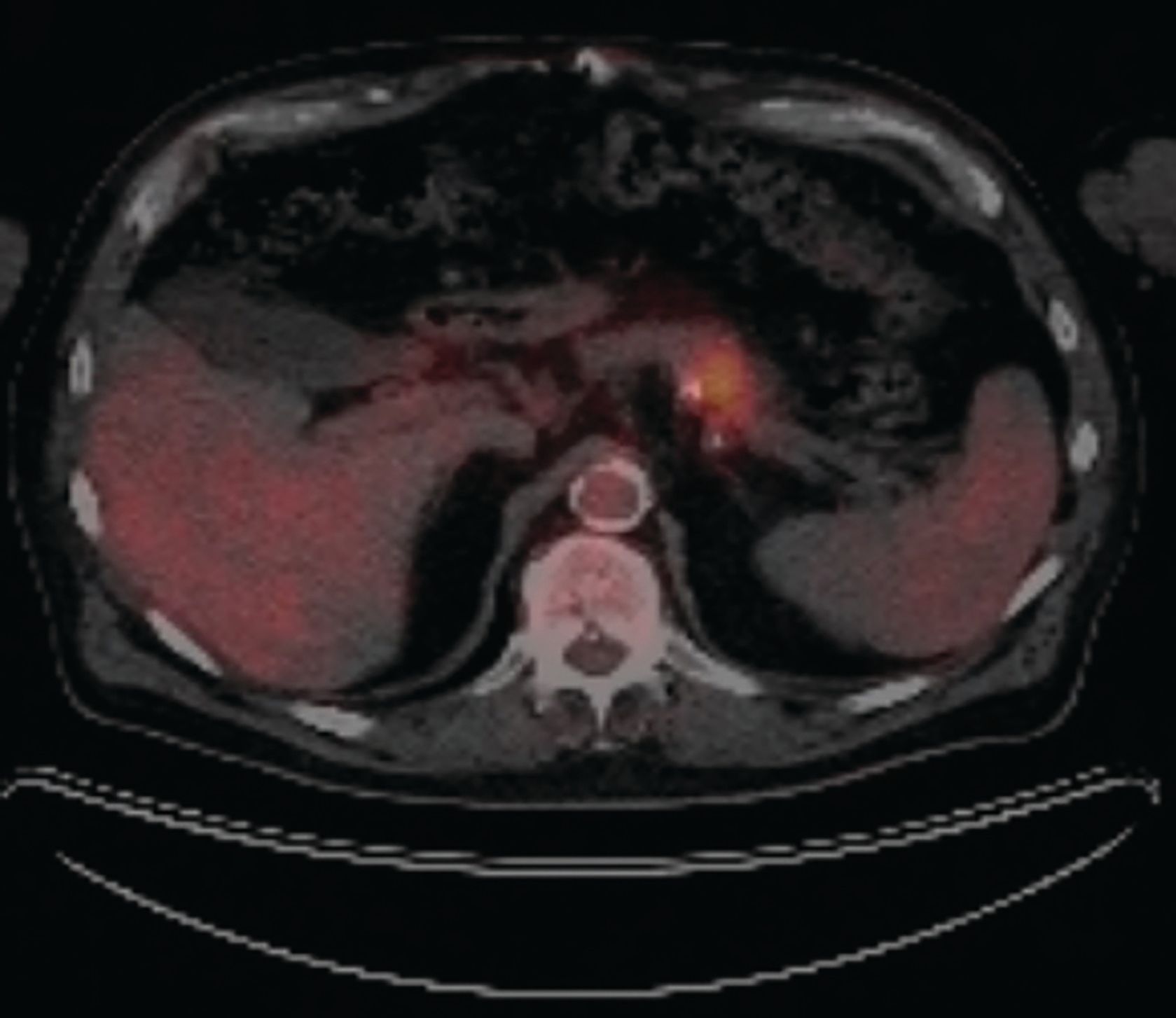
Figure 1. Positron emission tomography (PET) CT scan 8/1/19: small focus of increased fluorodeoxyglucose (FDG) activity within the pancreatic tail, suspicious for pancreatic malignancy measuring approximately 26 x 18 mm.
Case
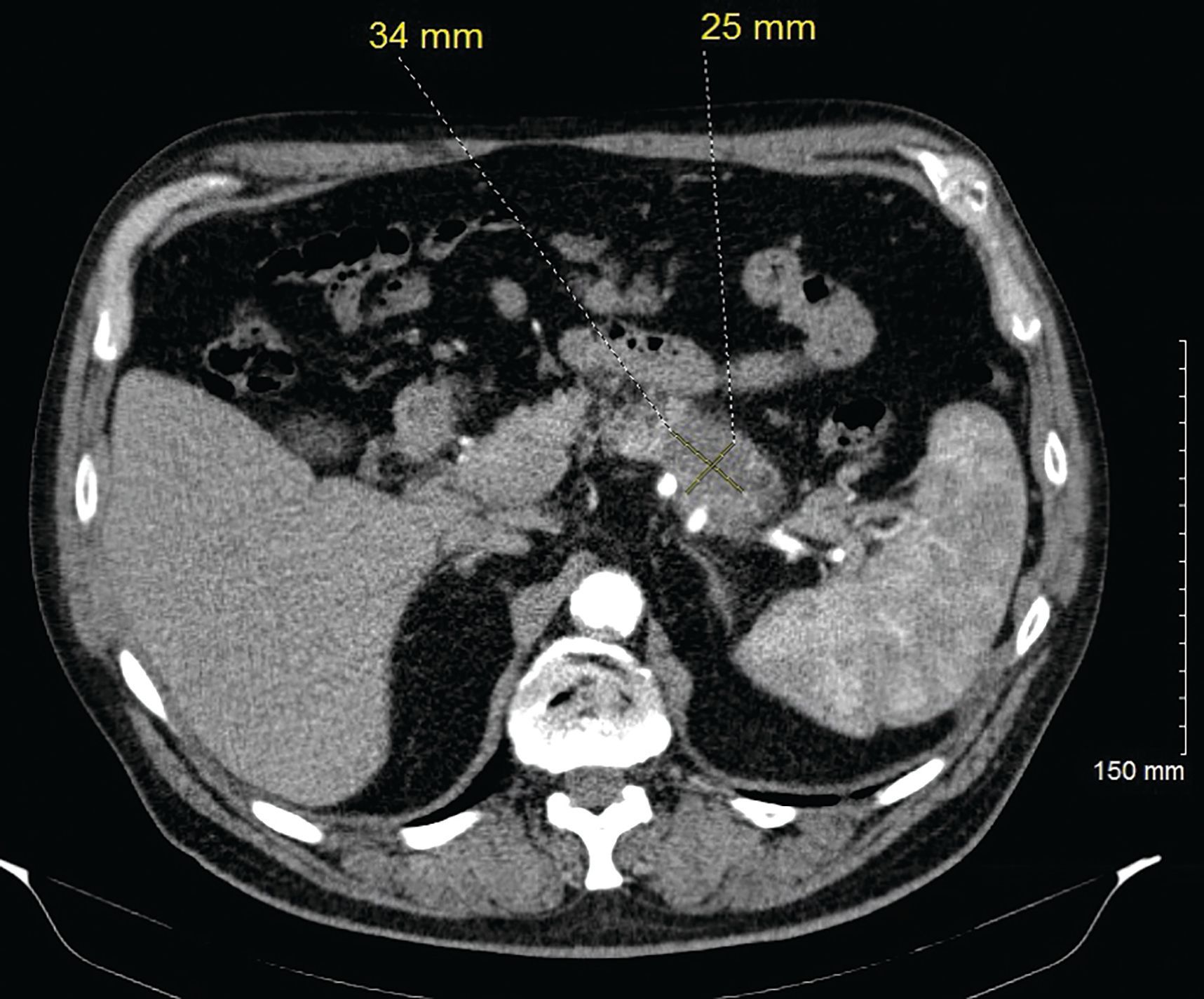
A Caucasian male, aged 74 years, was incidentally found to have a pancreatic mass during a surveillance imaging positron emission tomography (PET) scan for his prior malignant melanoma. The PET scan detected a suspicious uptake in the pancreatic tail corresponding to a subtle 2.6 cm x 1.8 cm region of low attenuation with a standardized uptake value of 7.5 (Figure 1). A CT scan of the abdomen revealed this to be a hypodense mass in the distal body/tail portion of the pancreas measuring 3.4 cm x 2.5 cm (Figure 2). Further work-up with upper endoscopy and endoscopic ultrasound biopsy revealed invasive PDAC. Family history was significant; his father and paternal grandfather had both received a diagnosis of PDAC around age 70 years and died from it. A genetic counseling visit and germline multigene hereditary pancreatic cancer panel testing detected no pathogenic sequence variants or deletions/duplications in the analyzed genes; these included APC, ATM, BRCA1, BRCA2, CDKN2A, MLH1, MSH2, MSH6, PALB2, PMS2, STK11, and TP53 (sequencing and deletion/duplication), and EPCAM (deletion/duplication only).


Figure 2. Axial contrast enhanced CT (arterial phase) demonstrates pancreatic tail lesion with relative hypoenhancement, corresponding to the focus of uptake on the recent positron emission tomography (PET) imaging. There was dilatation of the pancreatic duct distal to this finding (not shown), which did not show fluorodeoxyglucose (FDG) uptake on PET.
The medical history was significant for a diagnosis of malignant melanoma in his right lower extremity, diagnosed and treated with curative intent 3 years earlier. It had a Breslow’s depth of 1.45 mm, Clark’s level IV, and ulcerated superficial spreading. He was initially treated with wide local excision and a sentinel node procedure, which revealed 1 positive lymph node without extracapsular extension. Further surgery with radical superficial groin dissection showed no further evidence of melanoma. He was treated with adjuvant immunotherapy with nivolumab (Opdivo) for 1 year, and he currently had no evidence of malignant melanoma.


Figure 3. 12/6/19. Axial contrast enhanced CT (arterial phase) again demonstrates the subtle hypoenhancing lesion in the pancreatic tail. Decreased from 34 x 25 mm to 15 x12 mm.
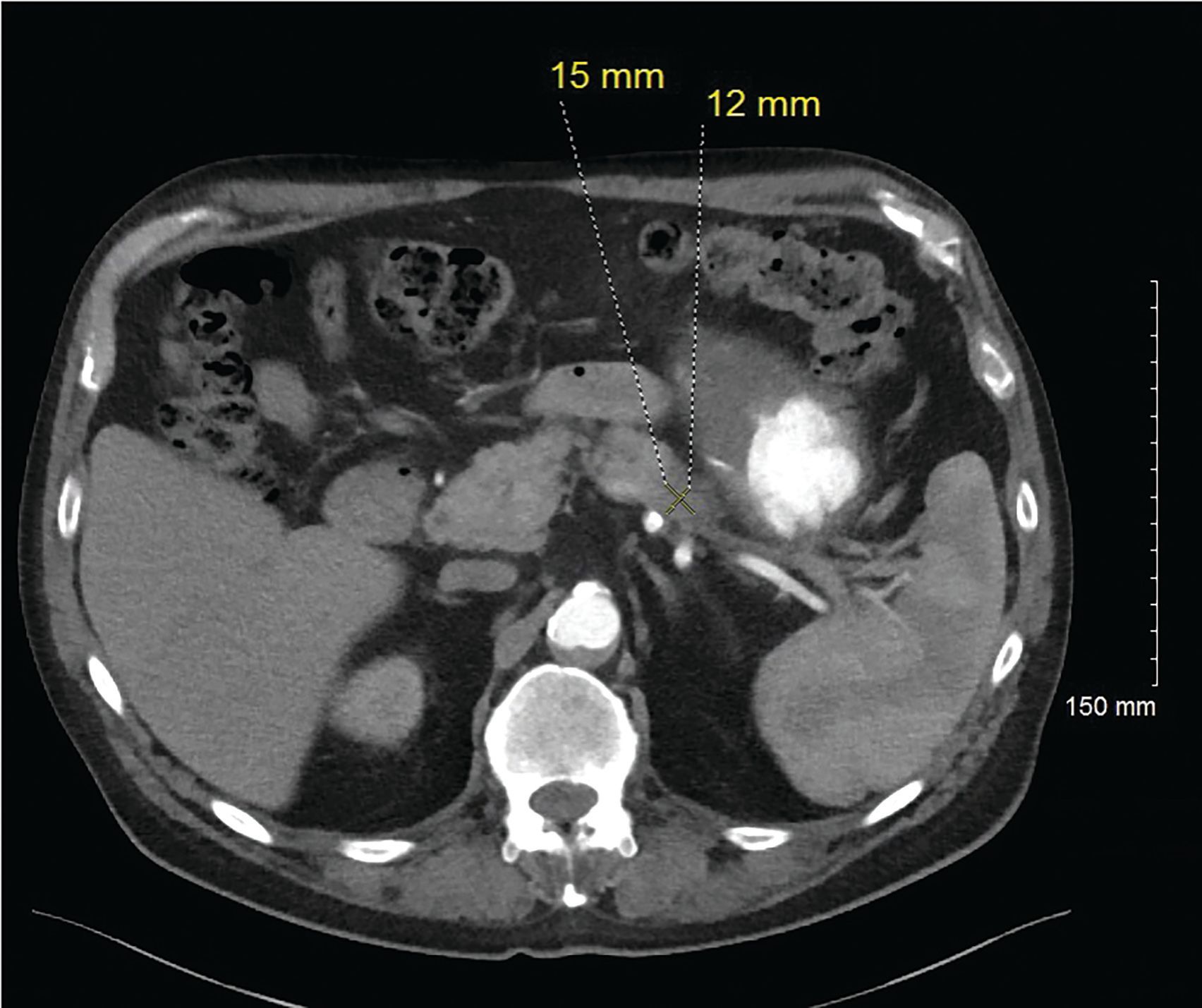
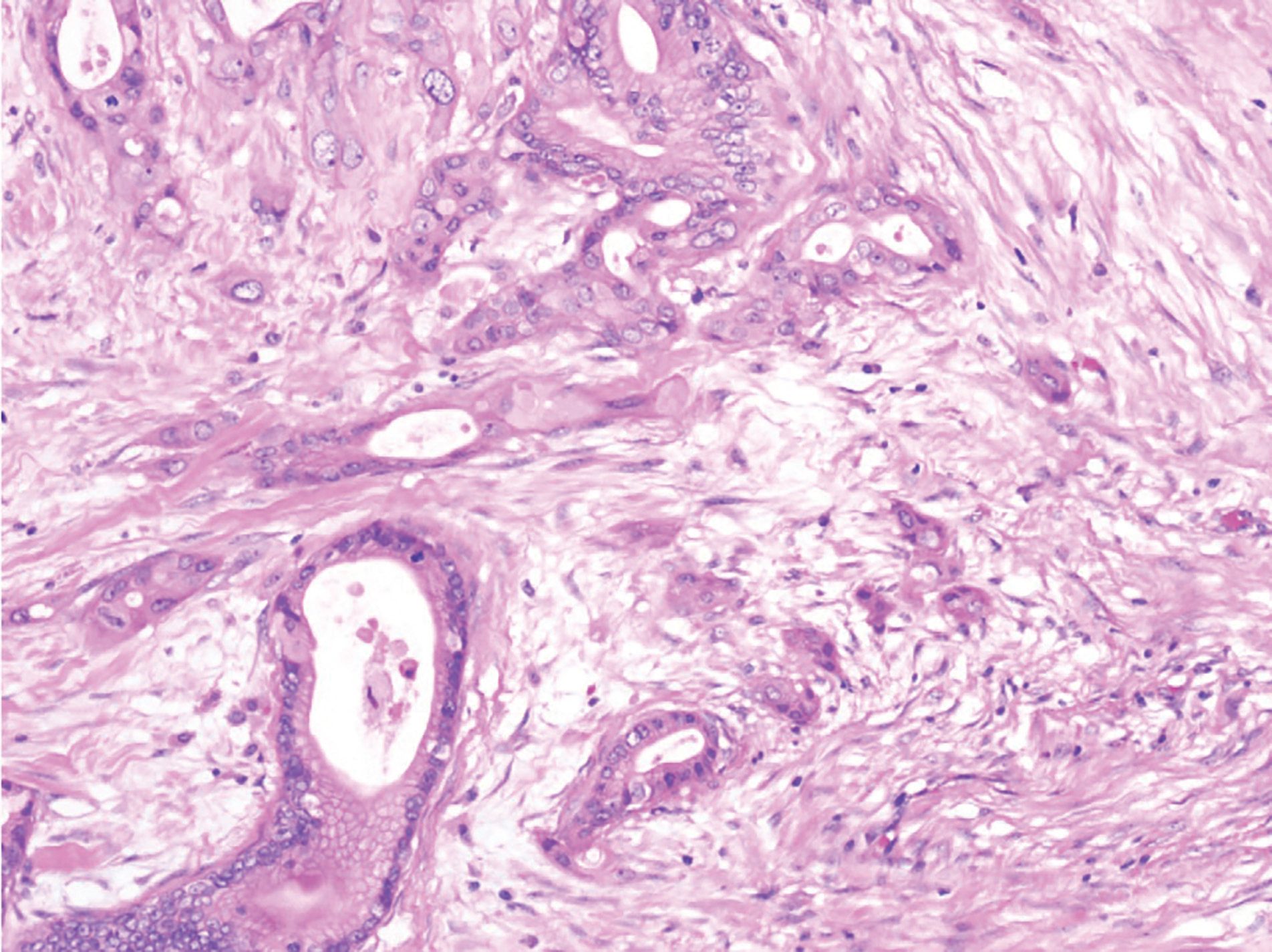
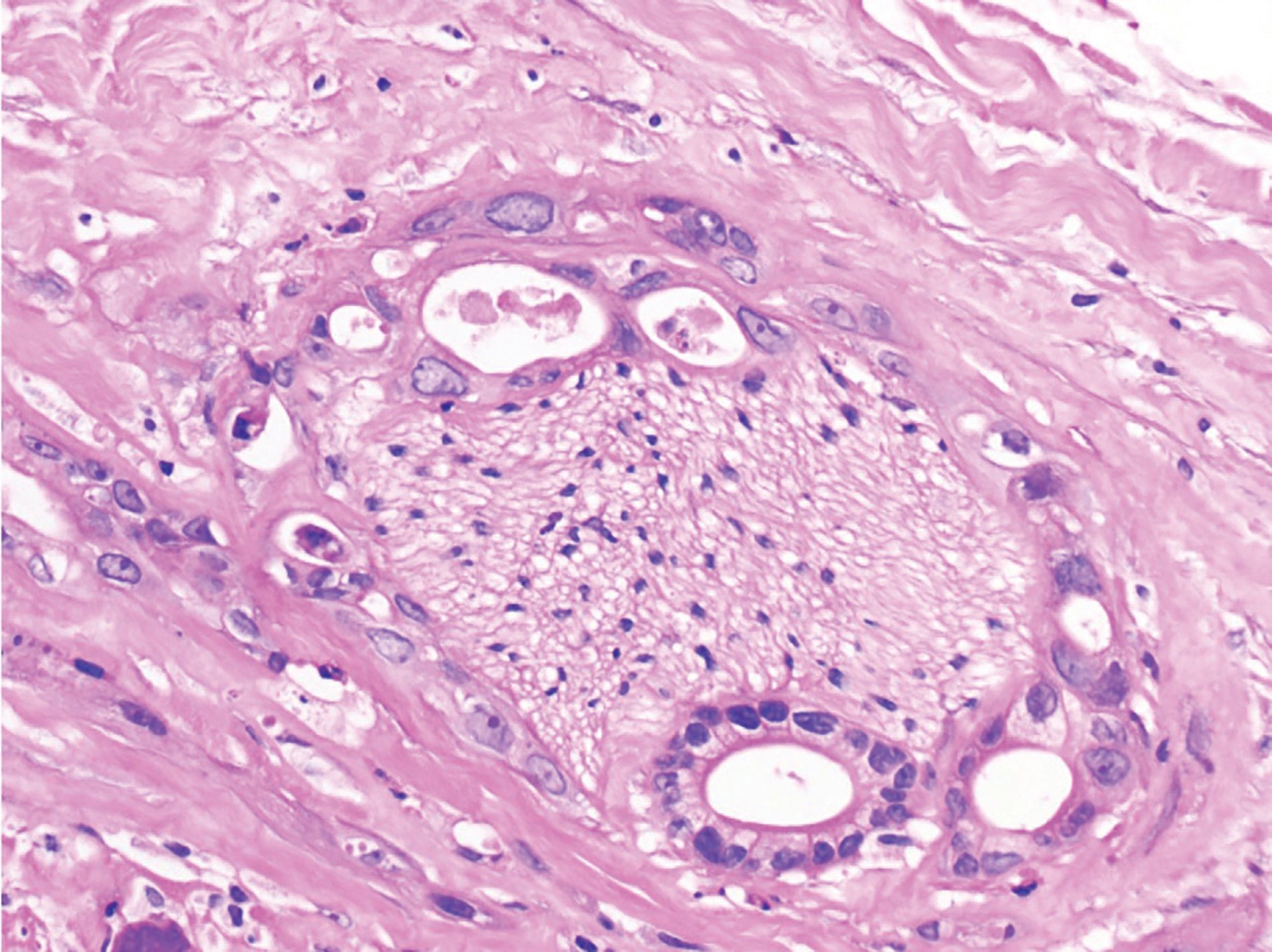
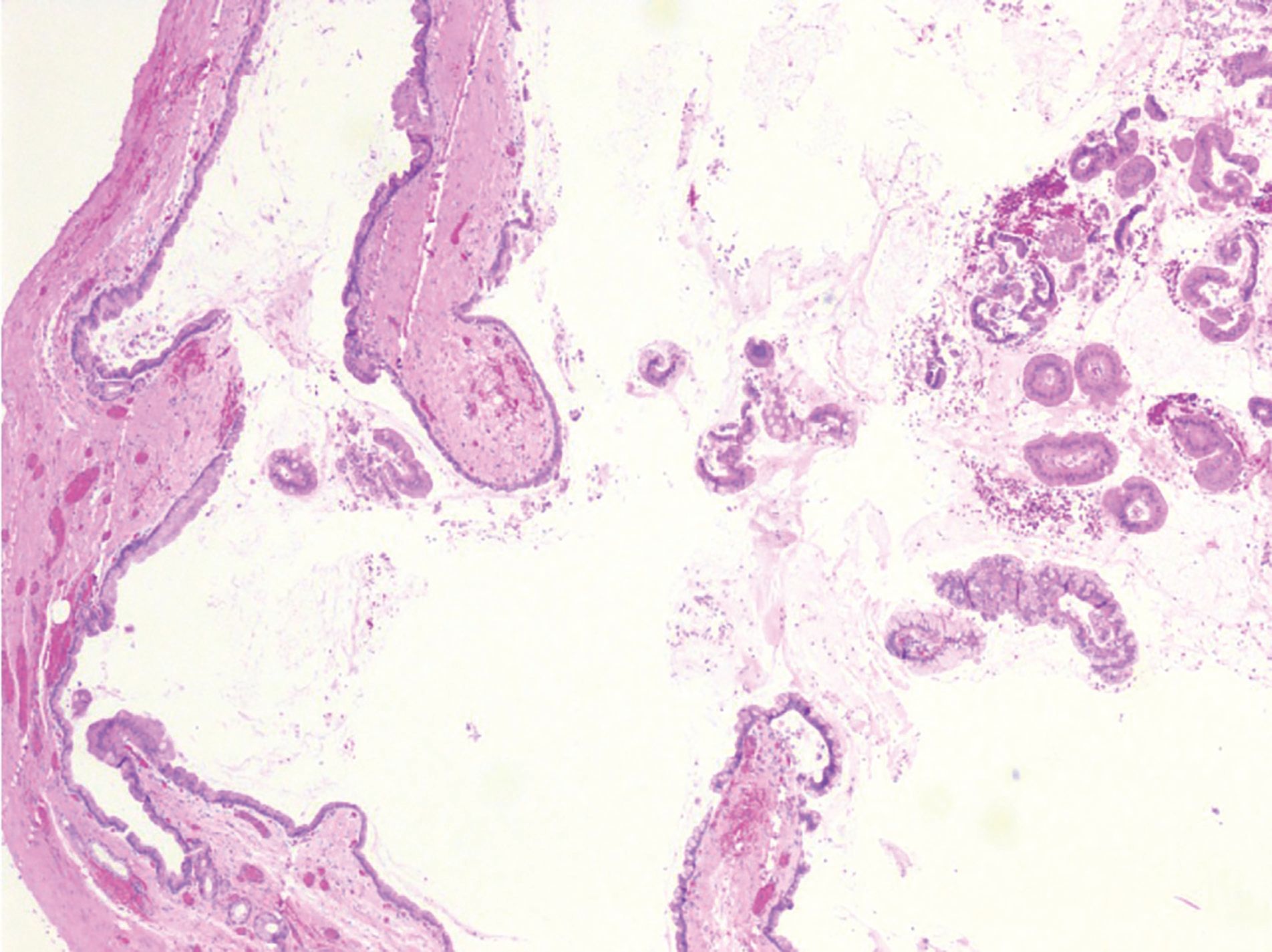
For his newly diagnosed locally advanced PDAC, the patient was treated with 6 cycles of a neoadjuvant FOLFIRINOX chemotherapy regimen, which reduced his pancreatic mass from 3.4 cm x 2.5 cm to 1.5 cm x 1.2 cm on imaging studies (Figure 3). He underwent a successful partial pancreatic body/tail pancreatectomy and splenectomy. Pathology revealed a 2.4-cm (pT2) residual invasive moderately differentiated PDAC arising in association with a low-grade intraductal papillary mucinous tumor at the proximal margin. While there was some perineal invasion, there was no lymphovascular invasion or any involved lymph nodes, of 14 removed (pN0). All resection margins were negative (R0 resection) (Figures 4, 5, 6). Spleen was without diagnostic abnormality. Immunostaining for DNA mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2) revealed retained nuclear staining for all 4 antigens. Results of the next-generation sequencing of tumor tissue revealed a microsatellite-stable tumor with a tumor mutational burden of 3 mutations per megabase. Genomic findings showed CDKN2A/B loss and presence of KRAS-Q61H, SGK1-K138fs13, SMAD4-P18fs17, and TP53-R273H, with no reportable therapeutic or clinical trial options.


Figure 4. Distal pancreatectomy showing invasive, moderately differentiated adenocarcinoma (H&E, 200X magnification).
Discussion
In most cases, the genetic basis of inherited PDAC is not well understood. Several large epidemiological studies have established the fact that a family history of pancreatic cancer increases the risk of developing the disease. However, as many as 80% of patients with a family history of pancreatic cancer have no identifiable genetic cause.27 A prospective registry study revealed that having 1 first-degree relative with PDAC increased the risk of developing the disease up to 2- to 5-fold, and having 2 first-degree relatives with the disease increased the risk to 6.4-fold.10 Early-onset (<50 years) pancreatic cancer in the family is associated with an even greater risk. The lifetime PDAC risk increases as the age of family members with the disease decreases.

Figure 5. Perineural invasion (H&E, 200X magnification).
Peutz-Jeghers syndrome is an autosomal dominant disorder characterized by hamartomatous polyps in the gastrointestinal tract; by pigmented macules of the lips, buccal mucosa, and digits; and increased risk of gastrointestinal cancers, including pancreatic cancer. Germline mutations of the STK11/LKB1 gene have been attributed to Peutz-Jeghers syndrome, which results in risk of PDAC increasing by as much as 132-fold. Inactivation of STK11/LKB1, by homozygous deletions or by somatic sequence mutations coupled with loss of heterozygosity, has been demonstrated in 4% to 6% of sporadic pancreatic cancer cases; this indicates a possible causal role in the carcinogenesis for both sporadic and inherited forms of PDAC.28

Figure 6. The tumor arose in association with a low-grade papillary mucinous neoplasm (H&E, 40X magnification).
Hereditary/familial pancreatitis syndrome is a rare cause of chronic pancreatic inflammation. Its onset is early, usually during childhood. It often starts with recurrent episodes of acute pancreatitis, and the clinical phenotype is not very different from other etiologies of the disease. The long-lasting inflammation generates a tumor-promoting environment predisposing to carcinogenesis. Several genes have been implicated with the familial form of pancreatitis, including PRSS1, SPINK1, and CFTR. The increased risk of developing PDAC has been estimated as between 26- and 87-fold.15,16,29,30
Familial malignant melanoma syndrome, also known as melanoma–pancreatic cancer syndrome or familial atypical multiple mole melanoma syndrome, is an autosomal dominant disease characterized by the familial occurrence of malignant melanoma of the skin and multiple atypical precursor lesions. Germline mutations in the p16 (CDKN2A) gene have been reported in at least a quarter of such families and associated with pancreatic cancer.31 In an analysis of 521 patients who met criteria for familial pancreatic cancer, 2.5% had germline mutations in CDKN2A, while patients with a family history of pancreatic cancer and melanoma had germline mutations of CDKN2A in 7.8% of cases.32 Familial malignant melanoma syndrome is associated with a 20- to 47-fold increased risk of pancreatic cancer.18 Patients with this syndrome have an earlier onset of pancreatic cancer compared with the general population.33
Lynch syndrome is an inherited cause of colorectal cancer caused by mutations of DNA mismatch repair genes MLH1, MSH2, MSH6, or PMS2. A number of extracolonic tumors have been associated with this disorder, including pancreatic cancer. The risk for pancreatic cancer can be as high as 9- to 11-fold greater than average in affected patients.34,35 Mutations in germline mismatch repair genes result in the lack of repair of errors that are introduced during DNA replication; these errors shorten or lengthen microsatellites, leading to their persistence on somatic cells. These can be tested on somatic tumor tissue samples. Microsatellite instability is also a prognostic factor for survival. The accumulated risk of pancreatic cancer in patients with Lynch Syndrome is around 3.7%. Pancreatic tumors developing in patients with Lynch Syndrome often present with a characteristic medullary appearance and prominent lymphocytic infiltration. Individuals with Lynch syndrome have an 8.6-fold increase in risk of pancreatic cancer development compared with the general population.36,37
Hereditary breast–ovarian cancer syndrome represents another genetic syndrome in which an excess of pancreatic incidence has been reported. In families harboring BRCA1/2 mutations, the risk of pancreatic cancer is increased by 2- to 6-fold, and the age of onset is younger than the average in the general population. BRCA1/2 mutations occur in 4% to 7% of all patients with pancreatic cancer.21,38,39
Familial pancreatic cancer is defined as having at least 2 first-degree relatives with pancreatic cancer that occurs in association with none of the genetic cancer syndromes described above. Although this syndrome does not seem to follow a specific Mendelian pattern of inheritance, research is under way to better understand this cohort of patients. Hereditary/familial syndromes associated with PDAC are listed in Table 2.
Because of the low incidence of PDAC in the general population, with a lifetime risk of 1.5%, screening is not feasible. It should, however, be considered in high-risk individuals, especially those with 5- to 10-fold or higher increased risk for PDAC. This scenario includes hereditary syndromes associated with increased risk of PDAC and members of families with familial pancreatic cancer. The purpose of screening is to detect precursor lesions or early cancer. Identification of PDAC at an early stage can be essential to improved survival, as demonstrated by the small proportion of patients with localized cancers who reach the 5-year survival mark at a rate of 31.5%. Moreover, recent data suggest that some specific germline mutations (mainly related to homologous repair) could be therapeutically targetable and guide personalized therapy. Identifying patients with predisposing genetic factors can improve clinical outcomes.
Outcome of the Case
The patient had a successful curative-intent treatment and currently has no evidence of cancer. His personal history of pancreatic cancer and malignant melanoma and a strong family history of pancreatic cancer—including 1 first-degree relative (father) and 1 second-degree relative (paternal grandfather)—put him and his family at an increased risk for future cancers, although that increased risk is not yet well defined. His current clinical presentation does not match any of the above-mentioned hereditary cancer syndromes. There seems to be a familial form of vertically inherited pancreatic cancer susceptibility in his family, despite the absence of clinically significant mutations in his germline multigene hereditary pancreatic cancer panel testing. Possible explanations for this presentation may include an alteration in a gene that cannot be currently detected with available technology, or there may be an entirely different, as-yet-undiscovered cancer-risk gene involved for which testing is not yet available. As no mutation was detected, his assessed risk to develop another cancer will be mostly based upon his medical and family history.
The patient’s first-degree relatives may remain at an elevated risk to develop pancreatic cancer. He was asked to notify us if any new cancers developed in his family, so we could assess if other testing would be appropriate. He and his family members were also encouraged to consider surveillance options and to potentially participate in screening, early detection, and prevention clinical trials.
Conclusions
Patients who receive a diagnosis of PDAC should undergo evaluation for hereditary syndromes known to be associated with increased risk for PDAC. Similarly, individuals with a family history of PDAC—whether they themselves are affected yet by cancer or not—who meet the criteria for familial pancreatic cancer, or those with 3 or more diagnoses of pancreatic cancer on the same side of their family, have an increased risk for pancreatic cancer and should be candidates for genetic testing, as should individuals who meet criteria for other genetic syndromes associated with increased risk for pancreatic cancer. Such genetic testing should include a comprehensive review of family history of cancer, preferentially with the help of a genetic counselor. Germline genetic testing for cancer susceptibility may be discussed with individuals with a pancreatic cancer diagnosis, even if family history is unremarkable. Benefits and limitations of pancreatic cancer screening should be discussed with individuals whose family history meets criteria for familial PDAC and/or genetic susceptibility to pancreatic cancer.
Financial Disclosure: The authors have no significant financial interest in or other relationship with the manufacturer of any product or provider of any service mentioned in this article.
References
1. Cancer facts & figures 2020. American Cancer Society. Accessed May 29, 2020. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html
2. Pancreatic cancer: statistics. Cancer.net. Accessed May 29, 2020 http://www.cancer.net/cancer-types/pancreatic-cancer/statistics
3. Arnold LD, Patel AV, Yan Y, et al. Are racial disparities in pancreatic cancer explained by smoking and overweight/obesity? Cancer Epidemiol Biomarkers Prev. 2009;18(9):2397-2405. doi:10.1158/1055-9965.EPI-09-0080
4. Cancer stat facts: pancreas cancer. National Cancer Institute/Surveillance, Epidemiology, and End Results Program. Accessed May29, 2020. https://seer.cancer.gov/statfacts/html/pancreas.html
5. Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393(4):535-545. doi:10.1007/s00423-007-0266-2
6. Lucenteforte E, La Vecchia C, Silverman D, et al. Alcohol consumption and pancreatic cancer; a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23(2):374-382. doi:10.1093/annonc/mdr120
7. Andersen DK, Andren-Sandberg Å, Duell EJ, et al. Pancreatitis-diabetes-pancreatic cancer: summary of an NIDDK-NCI workshop. Pancreas. 2013;42(8):1227-1237. doi:10.1097/MPA.0b013e3182a9ad9d
8. Bosetti C, Rosato V, Li D. Diabetes, antidiabetic medications, and pancreatic cancer risk: an analysis from the International Pancreatic Cancer Case-Control Consortium. Ann Oncol. 2014;25(10):2065-2072. doi:10.1093/annonc/mdu276
9. Hemminki K, Li X. Familial and second primary pancreatic cancers: a nationwide epidemiologic study from Sweden. Int J Cancer. 2003;103(4):525-530. doi:10.1002/ijc.10863
10.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64(7):2634-2638. doi:10.1158/0008-5472.can-03-3823
11. Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer. 2009;8(2):109-117. doi:10.1007/s10689-008-9214-8
12. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447-1453. doi:10.1053/gast.2000.20228
13. Whitcomb DC, Applebaum S, Martin SP. Hereditary pancreatitis and pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:201-209. doi:10.1111/j.1749-6632.1999.tb09524.x.
14. Rebours V, Boutron-Ruault M-C, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103(1):111-119. doi:10.1111/j.1572-0241.2007.01597.x.
15. Howes N, Lerch MM, Greenhalf W, et al; European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252-261. doi:10.1016/s1542-3565(04)00013-8
16. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89(x):442-446. doi:10.1093/jnci/89.6.442
17. Lynch HT, Fusaro RM, Lynch JF, Brand R. Pancreatic cancer and the FAMMM syndrome. Fam Cancer. 2008;7(1):103-112. doi:10.1007/s10689-007-9166-4
18. Vasen HF, Gruis NA, Frants RR, et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87(x):809-811.
19. Lynch HT, Deters CA, Lynch JF, Brand RE. Familial pancreatic carcinoma in Jews. Fam Cancer. 2004;3:233-240. doi:10.1007/s10689-004-9549-8
20. Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res. 2002;62(x):3789-3793.
21. Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310-1316. doi:10.1093/jnci/91.15.1310
22. Brose MS, Rebbeck TR, Calzone KA, et al. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365-1372. doi:10.1093/jnci/94.18.1365
23. Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790-1795. doi:10.1001/jama.2009.1529
24. Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81(2):214-218. doi:10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-I
25. Matsubayashi H, Takaori K, Morizane C, et al. Familial pancreatic cancer: concept, management and issues. World J Gastroenterol. 2017;23(6):935-948. doi:10.3748/wjg.v23.i6.935
26. Roberts NJ, Norris AL, Petersen GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6(2):166-175. doi:10.1158/2159-8290.CD-15-0402
27. Hruban RH, Canto MI, Goggins M, et al. Update on familial pancreatic cancer. Adv Surg. 2010;44:293-311. doi:10.1016/j.yasu.2010.05.011
28. Su GH, Hruban RH, Bansal RK, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers.Am J Pathol. 1999;154(6):1835-1840. doi:10.1016/S0002-9440(10)65440-5
29. LaRusch J, Solomon S, Whitcomb DC, et al. Pancreatitis overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, eds. GeneReviews. University of Washington, Seattle; 2014.
30. Rebours V, Lévy P, Ruszniewski P. An overview of hereditary pancreatitis. Dig Liver Dis. 2012;44(1):8-15. doi:10.1016/j.dld.2011.08.003
31. Whelan AJ, Bartsch D, Goodfellow PJ. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med. 1995;333(15):975-977. doi:10.1056/NEJM199510123331505
32. Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569-577. doi:10.1038/gim.2014.153
33. Lynch HT, Brand RE, Hogg D, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma–pancreatic carcinoma–prone families: the familial atypical mole melanoma–pancreatic carcinoma syndrome. Cancer. 2002;94(1):84-96. doi:10.1002/cncr.10159
34. Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790-1795. doi:10.1001/jama.2009.1529
35. Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30(9):958-964. doi:10.1200/JCO.2011.39.5590
36. Leoz ML, Sánchez A, Carballal S, et al. [Hereditary gastric and pancreatic cancer predisposition syndromes.]Gastroenterol Hepatol. 2016;39(7):481-493. doi:10.1016/j.gastrohep.2015.11.009
37. Banville N, Geraghty R, Fox E, et al. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum Pathol.2006;37(11):1498-1502. doi:10.1016/j.humpath.2006.06.024
38. Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutation in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95(3):214-221. doi:10.1093/jnci/95.3.214
39. Iqbal J, Ragone A, Lubinski J, et al; Hereditary Breast Cancer Study Group. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107(12):2005-2009. doi:10.1038/bjc.2012.483
















