Current Frontline Treatment of Multiple Myeloma
Sarah A. Holstein, MD, PhD, reviews updated treatment options for patients with newly diagnosed multiple myeloma.
ABSTRACT
Treatment paradigms for management of newly diagnosed (ND) multiple myeloma have been evolving over the past 20 years as a consequence of the development of immunomodulatory drugs, proteasome inhibitors, and monoclonal antibodies. While recent studies have continued to confirm the progression-free survival benefit of consolidation with upfront autologous stem cell transplant in those considered transplant eligible (TE), the line between induction strategies for TE and transplant-ineligible (TI) patients has blurred, based on studies evaluating both populations. Here, we present an overview of the data guiding current treatment approaches in the ND setting and discuss areas of ongoing investigation, including the role of quadruplet combination therapies in TE patients, the optimal strategies for frail TI patients, and management of high-risk disease.
Oncology (Williston Park). 2022;36(7):430-441.
DOI: 10.46883/2022.25920967
Introduction
The past 20 years have seen the development of immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and monoclonal antibodies (mAbs), which have dramatically reshaped the treatment paradigms for newly
diagnosed (ND) multiple myeloma (MM). While consolidation with high-dose melphalan and autologous stem cell transplant (ASCT) remains a standard of care (SOC) for patients who are considered to be transplant eligible (TE), the use of alkylating agents and other traditional cytotoxic agents has decreased dramatically. Effective triplet and even quadruplet combination therapies consisting of IMiDs, PIs, and/or mAbs can induce very deep responses, including achievement of minimal residual disease (MRD) negativity. These advances in therapies have led to considerable prolongation of survival times, although frail transplant-ineligible (TI) patients or patients with high-risk disease features continue to represent populations with inferior survival outcomes. In this review, we present an overview of the data that guide current treatment approaches for NDMM as well as the evolving strategies that may lead to future standards of care.
Current Standards for Diagnosis
Diagnostic evaluation for a patient suspected to have MM includes laboratory evaluation, a bone marrow aspirate/biopsy, and whole-body imaging. Laboratory evaluation should include a complete blood count with differential, comprehensive metabolic panel (including creatinine, calcium, and albumin), lactate dehydrogenase (LDH), β2-microglobulin, and testing for monoclonal proteins (including quantitative immunoglobulins, serum free light chains, serum and urine protein electrophoresis, and immunofixation). In addition to bone marrow aspirate and biopsy specimens sent for histopathological review, aspirate samples should also be sent for flow cytometric analysis to evaluate for the presence of clonal plasma cells as well as fluorescence in situ hybridization (FISH) on CD138-selected cells and cytogenetic testing. The Revised International Staging System (R-ISS) includes testing of albumin, β2-microglobulin, and LDH, as well as evaluation of key high-risk cytogenetic abnormalities observed from the FISH analysis of the bone marrow aspirate (including del[17p], t[4;14], and t[14;16]).1 Several options for whole-body imaging exist, including low-dose whole-body CT (LDWBCT), whole-body PET/CT, and whole-body MRI imaging.2 In both the International Myeloma Working Group and the National Comprehensive Cancer Network guidelines, the initial choices for imaging modality in the setting of suspected MM include either LDWBCT or PET/CT, with axial or whole-body MRI performed in the setting of negative or inconclusive testing.2,3 In the setting of known or suspected extramedullary involvement, PET/CT is the preferred imaging modality. These imaging modalities have replaced conventional skeletal surveys; the latter, while widely available, offer comparatively poor sensitivity. For example, in one head-to-head comparison between skeletal survey and LDWBCT, 25% of patients with negative skeletal surveys had lytic lesions on LDWBCT.4
Molecular Testing
Current staging and risk stratification systems primarily rely on information obtained from the CD138-selected FISH panel, which, at minimum, should evaluate for the presence of del(13), del(17p), t(4;14), t(11;14), t(14;16), and t(14;20), and for gain/amplification of 1q21 and del(1p). The frequency of these cytogenetic abnormalities is shown in Table 1. Within this panel, the “traditional” high-risk abnormalities have included del(17p), t(4;14), t(14;16), and t(14;20). Increasing evidence supports that 1q abnormalities, particularly amplification (>4 copies) or in combination with other high-risk abnormalities, should also be considered a high-risk abnormality.5 “Double-hit” biallelic inactivation of TP53 (eg, del[17p] in one allele plus mutation in the other allele) appears to be associated with the highest risk, although a recent publication demonstrated that del(17p) by itself is also associated with poor outcomes.6,7


TABLE 1. Frequencies of Cytogenetic Abnormalities in Multiple Myeloma
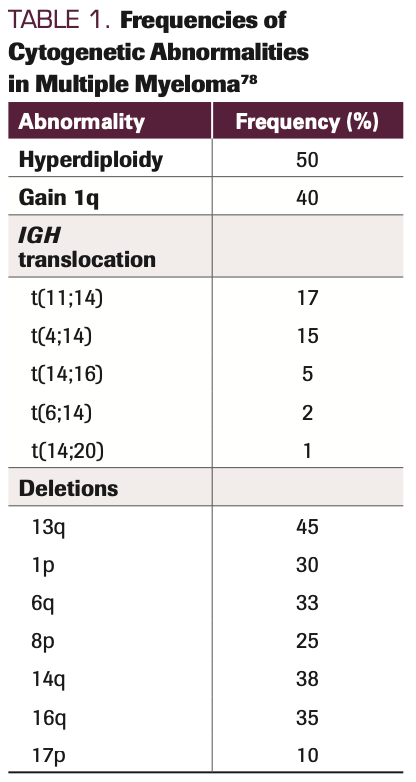
A number of gene signatures associated with standard- or high-risk disease have been reported over the years, and while the different panels have been demonstrated to identify different risk groups, these panels generally do not share any overlapping genes across the signatures.8 Thus, consensus has not been reached in the field regarding the optimal gene expression profiling (GEP) panel to utilize, and therefore this testing is not considered SOC.
Stratification for Treatment Selection
Transplant eligibility. For many patients, recommendations for initial treatment still hinge on whether the patient is considered to be eligible for high-dose therapy and ASCT. However, there is increasing overlap in the
induction strategies used in the transplant-eligible (TE) and transplant-ineligible (TI) groups, and multiple studies have demonstrated the feasibility and efficacy of ASCT in older adults.9-11 While ASCT candidacy is still sometimes in the eye of the beholder (namely, the transplant physician), several key factors that lead to ineligibility include poor performance status, liver cirrhosis, and heart failure (New York Heart Association functional status Class III/IV). Unfortunately, many patients who are potentially TE are never referred to an ASCT center for evaluation.12-15
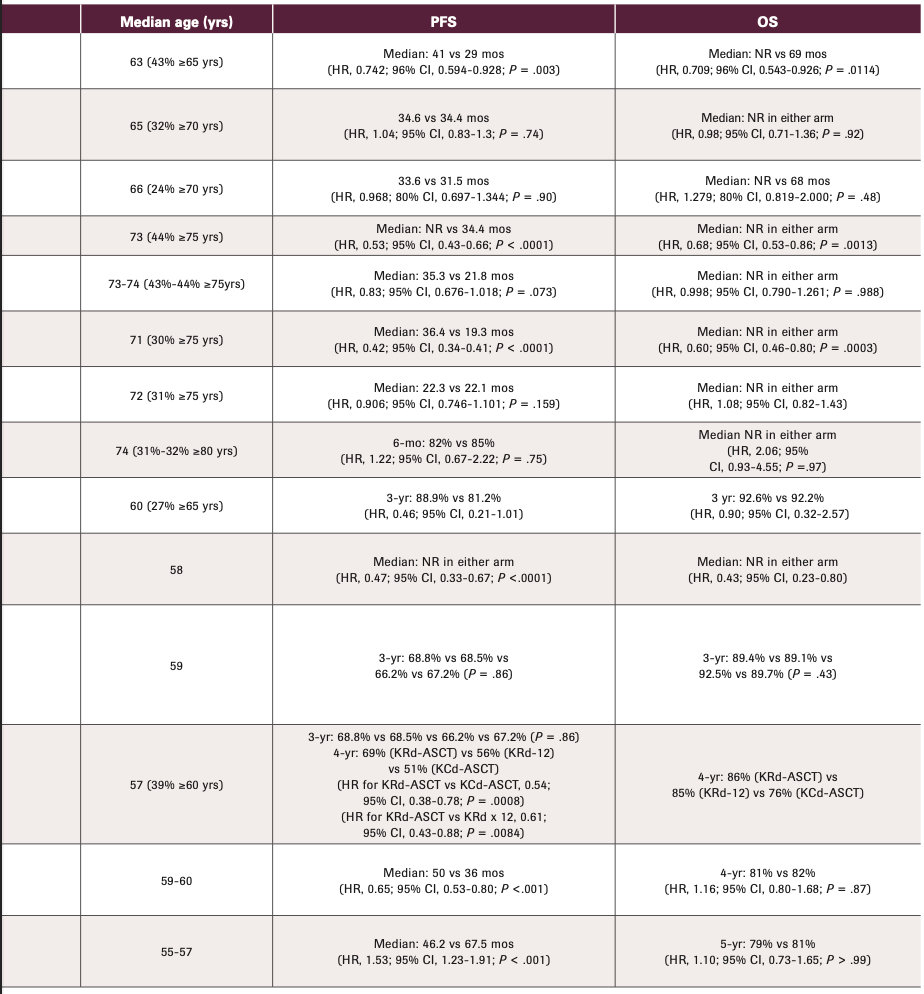
The line between TI and TE has also been blurred by the study design of several recent cooperative group phase 3 studies. In these, ND patients could either be TE who chose not to go through an upfront ASCT or TI (Table 2). As previously discussed,16 it can be difficult to apply the results of these studies (eg, SWOG S0777, ECOG E1A11) to the more typical TI patient seen in practice, one who is often older with more comorbidities. For instance, in the SWOG S0777 study (NCT00644228), 68% of the enrolled population were noted to have intent to pursue ASCT. Thus, while this trial is often used to support the assertion that lenalidomide (Revlimid), bortezomib (Velcade), and dexamethasone (RVd) is an SOC in the TI population, in actuality this study primarily enrolled a TE population who chose not to undergo upfront ASCT. In addition, the region of the world in which a trial was conducted is important, because age cutoffs for transplant eligibility may differ by nation. For example, in the MAIA study (NCT02252172; discussed further below), which helped establish the combination of daratumumab, (Dara; Darzalex), lenalidomide, and dexamethasone (Dara-Rd) as an SOC for TI patients, more than half of the enrolled subjects were aged less than 75 years17 and thus may have been considered TE in the United States.


TABLE 2. Summary of Recent Randomized Phase 2 or 3 Studies in Patients With Newly Diagnosed Multiple Myeloma
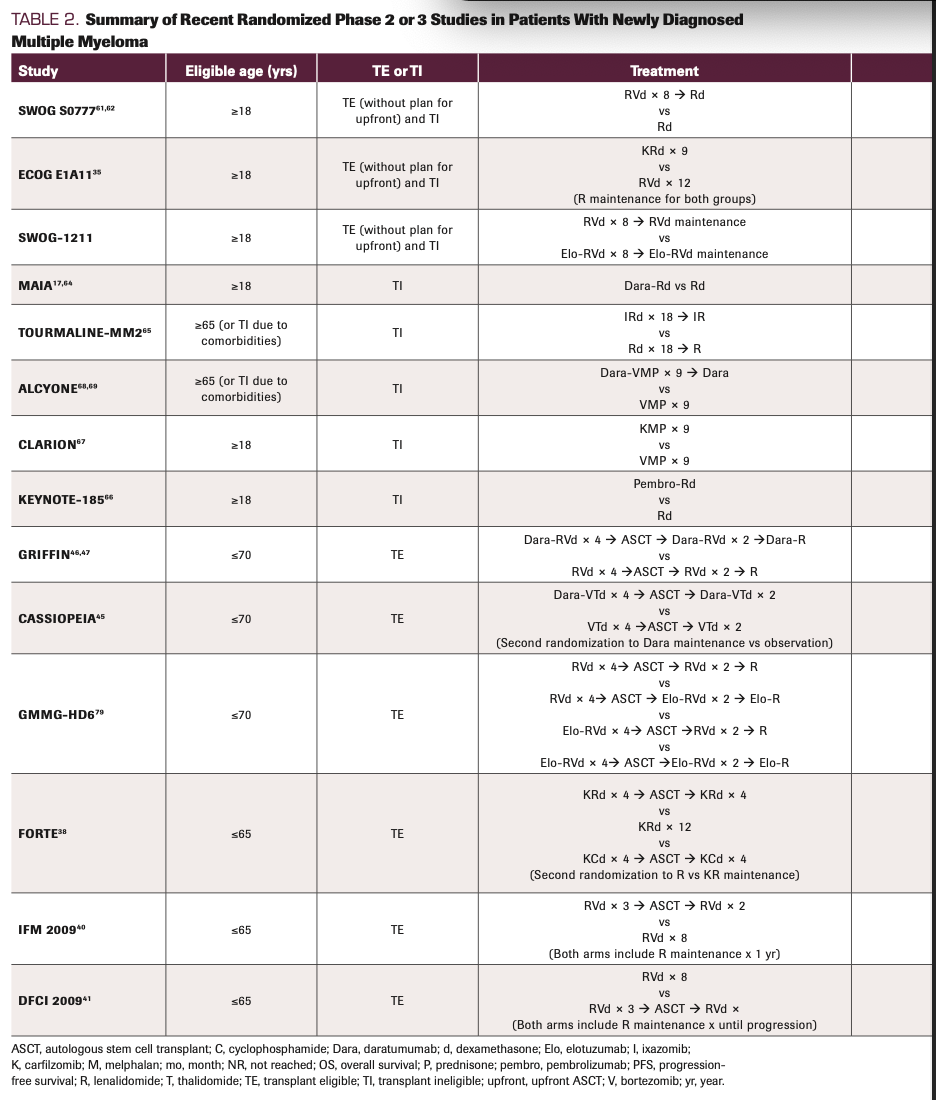


TABLE 2. Summary of Recent Randomized Phase 2 or 3 Studies in Patients With Newly Diagnosed Multiple Myeloma
Frailty-based assessment. A growing body of literature supports the use of geriatric and frailty assessment in determining treatment strategies for older adults who are being considered for ASCT as well as those who are considered TI.18-21 Frailty status has been shown to be a predictor of mortality in patients with MM.22,23 Multiple different frailty indices have been developed, including the International Myeloma Working Group (IMWG) Frailty Index, the Revised Myeloma Comorbidity Index, the Simplified Frailty Scale, and the UK Myeloma Research Alliance (UKMRA) Myeloma Risk Profile. The IMWG Frailty Index encompasses patient age, the Katz Activity of Daily Living scale, the Lawton Instrumental Activity of Daily Living scale, and the Charlson Comorbidity Index (CCI) score.24,25 The Revised Myeloma Comorbidity Index incorporates age, renal function, lung function, Karnofsky performance status, frailty (as determined by the Fried definition26), and disease cytogenetics.27 The Simplified Frailty Scale includes age, ECOG status, and CCI28 score; the UKMRA Myeloma Risk Profile includes age, World Health Organization performance status, ISS, and C-reactive protein.29
To date, the majority of the literature derives from post hoc analysis of clinical trials or large database reviews, and there is no consensus about the best available instrument to assess frailty.22,23,30-32 The Hovon 143 phase 2 study, which evaluated an induction strategy of daratumumab, ixazomib (Ninlaro), and dexamethasone, was of interest as it was specifically designed for frail TI patients.33 To be eligible for this study, patients had to be deemed frail by the IMWG frailty index. The median age of the enrolled subjects was 81 years (range, 70-92).33 While the reported overall response rate (ORR) was high (78%), the median progression-free survival (PFS) was only 13.8 months and 30% of patients died within the first year, highlighting the difficulty of treating this population. The ongoing FiTNEss (Frailty-adjusted therapy in Transplant Non-Eligible patients with newly diagnosed Multiple Myeloma) study (NCT03720041; Myeloma XIV) is a randomized phase 3 trial evaluating ixazomib/lenalidomide/dexamethasone (IRd) induction therapy in TE patients. The first randomization is to non–frailty-adjusted IRd induction vs frailty index-adjusted induction where different dose levels of lenalidomide and dexamethasone are used based on fit vs intermediate vs frail status.34 A second randomization involves maintenance with lenalidomide/placebo vs lenalidomide/ixazomib. The primary objectives are to compare early treatment cessation (<60 days from randomization) between standard and frailty-adjusted induction and PFS for the maintenance component. A report of the first 180 of a planned 740 subjects noted the feasibility of recruiting older patients (median age thus far 77 years, with 26% aged more than 80 years).34 Additional studies focused on these frail patients—who more accurately represent the real-world population of patients seen in practice—are needed.
Cytogenetic risk. Another potential stratification for treatment selection involves standard-risk vs high-risk disease. However, in the era of triplet/quadruplet induction regimens, few data suggest that one specific induction strategy has superior outcomes over others for patients with high-risk disease. This is primarily because the majority of randomized phase 2/3 studies have relied on underpowered subgroup analyses to compare standard- vs high-risk groups, and typically, patients with high-risk disease are underrepresented. This problem has been further exacerbated by differing definitions of high-risk disease (eg, whether 1q abnormalities are included, differing cut-offs for positivity, or exclusion of primary plasma cell leukemia) across studies.
The ENDURANCE trial (NCT01863550; ECOG E1A11) randomized patients to RVd vs carfilzomib (Kyprolis), lenalidomide, and dexamethasone (KRd) induction (without upfront ASCT) (Table 2).35 This study excluded patients with del(17p), t(14;16), t(14;20), LDH greater than 2 times the upper limit of normal (ULN), or greater than 20% circulating plasma cells. There was no difference in the primary end point (PFS) between the 2 treatment arms. Furthermore, subgroup analysis of the patients with t(4;14) did not reveal benefit for KRd over RVd (HR, 1.16; 95% CI, 0.54-2.47).35 In contrast, the SWOG-1211 study (NCT01668719) enrolled all of the high-risk patients who had been excluded from the ENDURANCE trial (high-risk GEP, t[14;16], t[14;20], del[17p], amp[1q], primary plasma cell leukemia, or LDH >2x ULN). In this study, which again did not include upfront ASCT, patients were randomized to elotuzumab (Empliciti) plus RVd vs RVd induction.36 This study failed to demonstrate benefit of adding elotuzumab to the backbone of RVd induction in patients with high-risk MM. The UK OPTIMUM/MUKnine trial (NCT03188172) exclusively enrolled patients considered to have ultra–high-risk disease, defined as having 2 or more high-risk features (t[4;14], t[14;16], t[14;20], gain[1q], del[1p], del[17p], gene expression SKY92 profiling, or >20% circulating plasma cells). This study was designed as a digital comparator-arm trial in which enrolled subjects were compared with molecularly matched patients from the National Cancer Research Institute Myeloma XI/XI+ trial (NCT01554852). A superior 18-month PFS rate (the primary end point) was observed with the UK OPTIMUM/MUKnine treatment strategy (involving a 5-drug induction regimen, transplant, and intensive
consolidation/maintenance strategies) compared with that achieved by the molecularly matched participants from the previous Myeloma XI trial: 81.7% vs 65.9%, respectively.37 It is hoped that ongoing phase 3 studies comparing quadruplets vs triplets (eg, PERSEUS [NCT03710603], GMMG HD7 [NCT03617731]) will provide more information regarding the potential benefit of quadruplets in patients with high-risk disease.
Therapy Selection
Transplant eligible. In the TE setting, induction therapy consisting of lenalidomide, a PI (either bortezomib or carfilzomib), and dexamethasone is established as SOC (Figure). A direct comparison of RVd and KRd induction followed by ASCT has not been conducted. As noted above, no difference in PFS (comparing RVd vs KRd) was observed in the ENDURANCE study; however, this study did not include consolidation with ASCT or most categories of high-risk disease.35 While prospective randomized data are lacking for the comparison of RVd vs the combination of bortezomib, cyclophosphamide, and dexamethasone (VCd), some trial results have demonstrated the superiority of bortezomib, thalidomide, and dexamethasone (VTd) over VCd (IFM2013-04; NCT01971658) as well as KRd over carfilzomib, cyclophosphamide, and dexamethasone (FORTE; NCT02203643).38,39


FIGURE. Current, Emerging, and Future Treatment Strategies for Newly Diagnosed Transplant- Eligible Multiple Myeloma

The IFM/DFCI2009 (NCT01191060) and FORTE trials demonstrated superior PFS rates for the inclusion of upfront ASCT as consolidation following RVd (IFM/DFCI2009) or KRd (FORTE) induction.38,40,41 None of these studies have demonstrated superior overall survival (OS) outcomes with inclusion of upfront ASCT, but given the increasing number of salvage therapies available and the overall trend for longer duration of survival in this patient population, it is becoming increasingly difficult to demonstrate OS benefit in the upfront setting.
As shown in Table 2 and Table 3, many recent and/or ongoing randomized studies have included post-ASCT consolidation with multiagent therapy prior to transitioning to maintenance therapy. Whether these cycles of post-ASCT consolidation contribute significantly to the long-term outcomes of these treatment strategies is not clear. The BMT CTN 0702 trial (NCT01109004) randomized patients who had completed induction therapy to 1 of 3 arms: single ASCT, tandem ASCT, or single ASCT followed by 4 cycles of RVd consolidation.42 All 3 arms then went on to receive lenalidomide maintenance. No differences in either PFS or OS outcomes were observed across the 3 arms, suggesting that single ASCT followed by lenalidomide maintenance was an SOC.42 The EMN02/HOVON 95 MM trial (NCT01208766) compared post-VCd induction therapy of the combination of bortezomib, melphalan, and prednisone (VMP) vs ASCT (1 or 2 transplants).43 A second randomization involved additional consolidation with either 2 cycles of RVd or nothing, followed by lenalidomide maintenance in both arms. As a whole, a superior PFS rate was observed for those receiving RVd consolidation (59.3 vs 42.9 months; HR, 0.81; 95% CI, 0.68-0.96; P = .016).44 However, upon closer examination of the subset of patients who received ASCT, one observes that the trend toward superior PFS with RVd consolidation did not quite reach statistical significance (HR, 0.83; 95% CI, 0.66-1.03).44 Of note, no patients in the EMN02/HOVON 95 MM trial received lenalidomide with induction, whereas 56% of patients in the BMT CTN 0702 trial received RVd induction and 10% received lenalidomide and dexamethasone (Rd) induction. Overall, outside the context of a clinical trial, patients commonly proceed directly from ASCT to maintenance therapy, bypassing multiagent consolidation.


TABLE 3. Ongoing Randomized Phase 3 Studies in NDMM
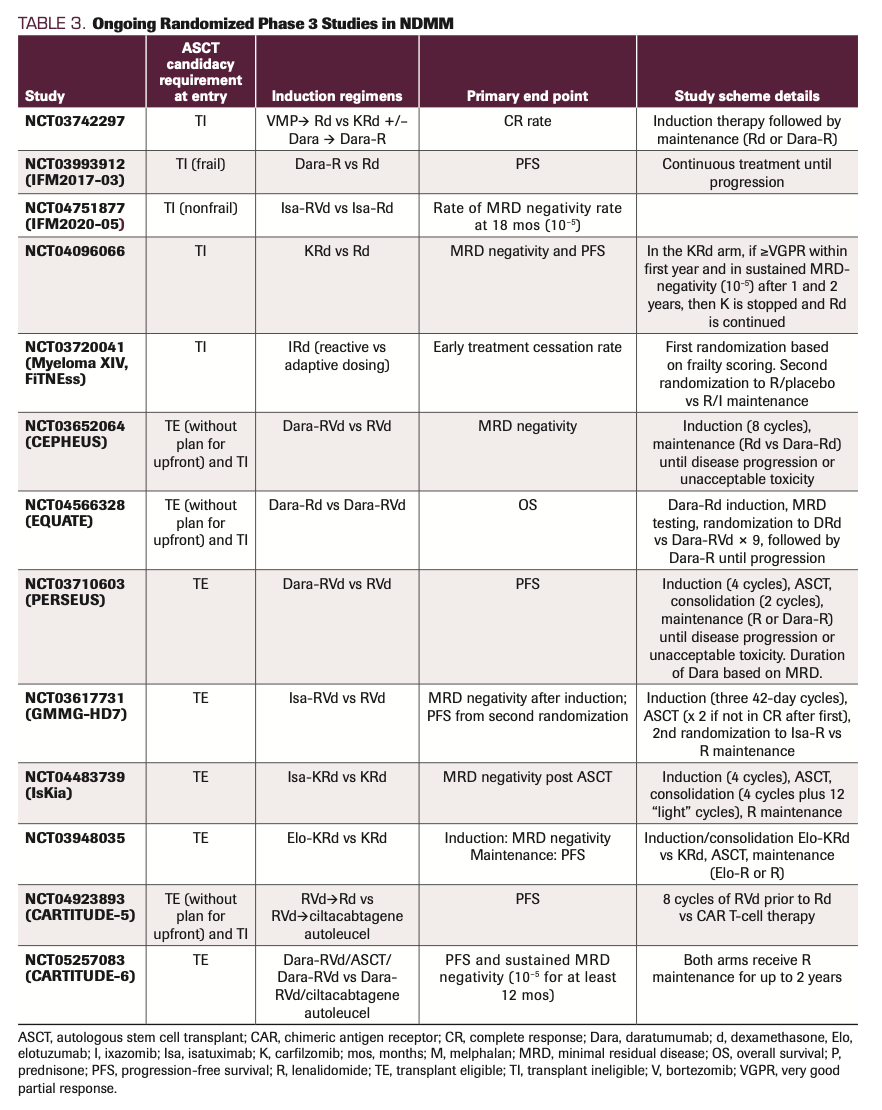
A major unanswered question in the TE setting is the potential role of mAb therapy as an addition to the backbone of IMiD/PI/dexamethasone induction; many recent and ongoing studies are addressing this knowledge gap (Tables 2 and 3). For instance, the CASSIOPEIA trial (NCT02541383), a randomized phase 3 study, evaluated the addition of daratumumab to the backbone of VTd induction/consolidation (Table 2). While the study met its primary end point (median PFS not reached in either arm; HR, 0.47; 95% CI, 0.33-0.67; P <.0001),45 resulting in the FDA approval of Dara-VTd, it has not been practice-changing in the United States given the use of thalidomide. Of more interest have been the studies evaluating the addition of mAb therapy to RVd or KRd backbones. GRIFFIN (NCT02874742), a randomized phase 2 trial, compared RVd induction, ASCT, RVd consolidation, and R maintenance with Dara-RVd induction, ASCT, Dara-RVd consolidation, and Dara-R maintenance. The study met its primary end point of stringent complete response (sCR) rate post consolidation (42.4% vs 32.0%; odds ratio [OR], 1.57; 95% CI, 0.87-2.82; 1-sided P = .068 with a prespecified 1-sided α of 0.10).46 However, the 24-month PFS rates were similar (95.8% vs 89.8%). With longer follow-up, the PFS curves have had more separation, not yet meeting statistical significance (3-year PFS rate, 88.9% vs 81.2%; HR, 0.46; 95% CI, 0.21-1.01).47 Higher rates of grade 3/4 neutropenia, thrombocytopenia, and upper respiratory tract infections were observed in the daratumumab-containing group.47 The ongoing phase 3 PERSEUS study (Table 3), which has a similar design (with the exception of the maintenance portion), is powered to evaluate PFS as the primary end point and should therefore provide more substantive evidence for the use of Dara-RVd as an induction strategy. The phase 3 GMMG-HD7 trial evaluated isatuximab (Isa; Sarclisa), another anti-CD38 mAb, in the context of RVd induction. This study was designed to evaluate the achievement of MRD negativity (at 10–5 sensitivity using flow cytometry) post induction therapy as the primary end point. A statistically significant improvement in rates of MRD negativity post induction was observed for the Isa-RVd arm (50.1%) vs the RVd arm (35.6%) (OR, 1.83; 95% CI, 1.34-2.51; P <.001).48 No data regarding PFS outcomes have been reported thus far. Several single-arm studies have evaluated Dara-KRd induction (with or without ASCT) and reported high rates of MRD negativity.49,50 As neither sCR nor rates of MRD negativity have been confirmed as surrogate end points for PFS or OS, further follow-up is required from ongoing studies to determine whether initial achievement of deeper response rates in the setting of quadruplet therapies translate to superior long-term outcomes.
An in-depth discussion of maintenance is outside the scope of this review and is discussed more extensively elsewhere.51-53 However, the current SOC, established through four randomized phase 3 studies, is the use of single-agent lenalidomide maintenance therapy. These studies demonstrated significant PFS benefit with lenalidomide maintenance (compared with placebo or observation) and in aggregate also showed superior OS outcomes.54-59 Current unanswered questions include whether addition of mAb therapy to the backbone of lenalidomide maintenance improves outcomes, whether maintenance must be continued indefinitely to achieve optimal long-term outcomes, and whether a response-adapted approach using MRD negativity can be utilized to determine maintenance intensity and duration.
Transplant ineligible. Many of the randomized studies conducted over the past decade have built upon the backbone of continuous Rd induction, which had previously been established as an SOC by the FIRST trial (NCT00689936).60 As noted above, SWOG S0777 randomized patients to RVd vs Rd induction in the absence of ASCT, but the minority of enrolled subjects were considered TI.61 This study demonstrated superior PFS and OS outcomes with the triplet vs the doublet (Table 2).61,62 However, it is again worth noting that the majority of patients enrolled in this study were TE. As real-world experience has highlighted the difficulties of utilizing full-dose lenalidomide, twice-weekly bortezomib, and/or prolonged dexamethasone in more frail patients, a single-arm phase 2 study was conducted to evaluate the “RVd lite” regimen.63 In this study, lenalidomide was decreased to 15 mg, bortezomib was administered once weekly (subcutaneously), and dexamethasone was administered only during the induction phase. The trial consisted of 9 cycles of triplet therapy followed by 6 cycles of consolidation with lenalidomide and bortezomib.63 Maintenance was not dictated by the trial, but lenalidomide maintenance could be utilized per investigator discretion. Consistent with a TI population, the median age was 73 years (range, 65-91). The reported median PFS was 35.1 months (95% CI, 30.9 to not reached).63 The overall feasibility of this approach was demonstrated by 64% of the patients completing the 15 cycles of induction/consolidation (with only 4% discontinuing due to toxicity) and patient-reported quality-of-life outcomes showing statistically significant improvements in physical function, future perspective, and disease symptom scores at the end of treatment compared with baseline.63
MAIA, a phase 3 study comparing Dara-Rd with Rd in the ND TI setting, has established Dara-Rd as an SOC in that setting. With a median follow-up of 56.2 months, the median PFS was not reached in the Dara-Rd group, compared with 34.4 months in the Rd group (HR, 0.53; 95% CI, 0.43-0.66;
P <.0001).64 Furthermore, an OS benefit was observed (HR, 0.68; 95% CI, 0.53-0.86; P = .0013).64 Median duration of response, overall response rate, and rate of MRD negativity were all superior in the Dara-Rd arm.64
The addition of a third drug to the Rd backbone has not always resulted in superior outcomes. In the phase 3 TOURMALINE-MM2 study (NCT01850524), patients were randomized to the combination of ixazomib, lenalidomide, and dexamethasone (IRd) vs Rd.65 While the median PFS of the IRd group improved numerically relative to the Rd group (35.3 vs 21.8 months), this did not meet statistical significance (HR, 0.83; 95% CI, 0.676-1.018; P = .073). No difference in OS was observed, although the median time to progression was superior in the IRd arm (45.8 vs 26.8 months; HR, 0.738; 95% CI, 0.589-0.925; P = .008).65 The phase 3 KEYNOTE-185 trial (NCT02579863) evaluated the addition of the immune checkpoint inhibitor pembrolizumab (Pembro; Keytruda) to the Rd backbone.61 Notably, this study, as well as several others involving combination therapy with checkpoint inhibitors and IMiDs, were halted by the FDA due to imbalances in the proportion of deaths. With short follow-up (median, 6.6 months), the 6-mos PFS rate estimates were 82.0% (Pembro-Rd) vs 85.0% (Rd; HR, 1.22; 95% CI, 0.67-2.22; P = .75). Overall response rates did not differ between the 2 treatment groups (64% vs 62%). Higher rates of serious adverse events (AEs) were observed in the Pembro-Rd arm (54%) compared with the Rd arm (39%) as were more treatment-related deaths (6 vs 1).66
VMP was historically an SOC induction regimen, particularly in Europe. The phase 3 CLARION study (NCT01818752) compared fixed-duration VMP induction therapy with carfilzomib, melphalan, and prednisone
(KMP). No differences in PFS (the primary end point), OS, complete response, or rate of MRD negativity were observed.67 Consistent with the known AE profiles of the 2 PIs, the VMP arm was associated with higher rates of peripheral neuropathy while the KMP arm was associated with higher rates of acute renal failure and cardiac failure.67 The results of this study, as well as those of ENDURANCE,35 failed to demonstrate superiority of carfilzomib over bortezomib in the ND setting.
The phase 3 ALCYONE trial (NCT02195479) built upon the VMP backbone by adding daratumumab.68,69 Fixed-duration VMP induction followed by observation was compared with Dara-VMP induction followed by daratumumab maintenance until progression. Here, the daratumumab-containing arm had significantly improved PFS (median, 36.4 vs 19.3 months; HR, 0.42; 95% CI, 0.34-0.41; P <.0001).68 Not surprisingly, there was more marked separation of the curves following the induction period, as the control arm was simply being observed off therapy (vs continued daratumumab therapy in the experimental arm). While neither the CLARION nor ALCYONE studies are particularly relevant to US ND TI treatment practices, given the backbones of melphalan plus prednisone, they do provide insight into the toxicity profile of upfront carfilzomib and the potential role for single-agent daratumumab maintenance, respectively.
Emerging and Novel Therapies
As noted above, the incorporation of anti-CD38 mAb therapy is now well established in the TI setting and likely represents a future SOC in the TE setting as well. However, there is also interest in the incorporation of other novel agents into the newly diagnosed setting, such as anti–B-cell maturation antigen (BCMA) agents, chimeric antigen receptor (CAR) T-cell therapy, the XPO1 inhibitor kselinexor (Seli; Xpovio), and cereblon E3 ligase modulators (CELMoDs). The first-in-class anti-BCMA antibody-drug conjugate (ADC) belantamab mafodotin (Bela; Blenrep) is currently approved as a single agent in the heavily relapsed/refractory setting. Several ongoing studies are evaluating Bela in combination with other MM therapies in earlier lines of therapy, including in the ND setting. A phase 2 study (NCT04802356) is evaluating the combination of Bela-RVd induction followed by ASCT, Bela-RVd consolidation, and then maintenance with Bela-R (up to 2 years of Bela; lenalidomide until progression). The Bela-RVd combination is also being studied in the ND TI setting (DREAMM-9, NCT04091126) in which patients receive 8 cycles of Bela-RVd followed by Bela-Rd until progression.70 During the phase 1 portion, the optimal dosing/schedule of Bela will be determined, followed by a phase 3 portion in which patients will be randomized 1:1 to Bela-RVd vs RVd. Determining a dose/schedule that minimizes the corneal toxicity associated with Bela, and thus allows long-term administration of this agent, will be critical. Also, some studies under development will incorporate novel bispecific antibody (BiAb) therapies such as teclistamab (an anti-BCMA/anti-CD3 agent) into the ND setting.
Several ongoing studies are evaluating the incorporation of BCMA-directed CAR T-cell therapy in the ND setting. The phase 1 KarMMa-4 (NCT04196491) study involves consolidation with idecabtagene vicleucel following induction (RVd, Dara-RVd, KRd, Dara-KRd, or CyBorD allowed) in patients with high-risk disease. Maintenance therapy with lenalidomide is allowed. The phase 3 CARTITUDE-5 (NCT04923893) trial randomizes patients who are not planning upfront ASCT to either RVd induction followed by ciltacabtagene autoleucel CAR T-cell therapy or RVd induction followed by Rd. In CARTITUDE-6 (NCT05257083), ND patients are randomized to treatment with Dara-RVd, ASCT, Dara-RVd, and R maintenance vs treatment with Dara-RVd, ciltacabtagene autoleucel (Carvykti), and R maintenance. The phase 2 BMTCTN1902 trial (NCT05032820) involves patients who have achieved less than a VGPR on lenalidomide maintenance within 12 months of ASCT; they will receive idecabtagene vicleucel (Abecma) CAR T-cell therapy followed by lenalidomide maintenance. Setting aside potential differences in cost, it remains to be seen whether CAR T-cell therapy will eventually replace ASCT
because of its ability to induce longer-lasting disease-free intervals.
Several studies are exploring the incorporation of selinexor into the ND setting. The preliminary reported results of patients enrolled in the Seli-Rd arm of the STOMP study (NCT02343042) include an ORR of 100% in 8 patients.71 An ongoing phase 2 study (NCT04782687) is exploring the combination of Seli-Dara-Rd in TI patients. Establishing a dose of Seli that has long-term tolerability will be critical, as use of this drug is often limited by its AE profile, which includes asthenia, weight loss, nausea, vomiting, and other gastrointestinal toxicity.72
Finally, the next-generation IMiD agents such as iberdomide and mezigdomide (CC-92480)—the CELMoDs—have shown promising activity in relapsed/refractory disease, including IMiD-refractory disease.73,74 Multiple ongoing studies are evaluating CELMoDs in combination with other anti-MM agents, such as proteasome inhibitors and alkylators. Several studies are incorporating iberdomide into the TI and TE ND settings (eg, NCT05272826, NCT05199311) and post-ASCT maintenance setting (eg, NCT05177536). If these agents, with their increased potency, are determined to have superior toxicity profiles, then they could take the place of currently used IMiDs (Figure).
Conclusions
Remarkable strides have been made in the treatment of NDMM in large part due to the development of IMiDs, PIs, and mAbs. Incorporation of anti-CD38 mAb therapy into the ND setting has been a significant advance for the TI population and appears poised to become an SOC in the TE setting as well (Figure). Looking ahead to the next 15 years, the hope is that incorporation of other strategies, including CAR T-cell therapy and BiAbs, will shift the treatment paradigms, potentially eradicating the disease with frontline therapy. Even if a cure remains out of reach during that timeframe, it is anticipated that incorporating novel therapies into the frontline setting will significantly improve survival outcomes. Whether these new therapies could be accompanied by prolonged treatment-free intervals, which would represent a notable shift from current treatment strategies, remains to be determined. Integral in the development of these emerging treatment paradigms will be incorporation of novel surrogate end points, such as MRD negativity, into trial design; this will not only allow earlier readouts of trials, but also help guide decision making regarding treatment de-escalation, intensification, or cessation.49,75-77
Disclosure: The author has served as a consultant for Bristol Myers Squibb/Celgene, GSK plc, Janssen Pharmaceuticals, Oncopeptides, Pfizer, Sanofi, Secura Bio, and Takeda Pharmaceutical Company; and has received research funding from Oncopeptides.
Corresponding author:
Sarah A. Holstein, MD, PhD
Division of Oncology and Hematology
Department of Internal Medicine
University of Nebraska Medical Center
Omaha, NE 68198
Phone: 402-559-8500
Fax: 402-559-6520
Email: sarah.holstein@unmc.edu
REFERENCES
- Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869. doi:10.1200/JCO.2015.61.2267
- Hillengass J, Usmani S, Rajkumar SV, et al. International Myeloma Working Group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019;20(6):e302-e312. doi:10.1016/S1470-2045(19)30309-2
- Callander NS, Baljevic M, Adekola K, et al. NCCN Guidelines Insights: multiple myeloma, version 3.2022. J Natl Compr Canc Netw. 2022;20(1):8-19. doi:10.6004/jnccn.2022.0002
- Hillengass J, Moulopoulos LA, Delorme S, et al. Whole-body computed tomography versus conventional skeletal survey in patients with multiple myeloma: a study of the International Myeloma Working Group. Blood Cancer J. 2017;7(8):e599. doi:10.1038/bcj.2017.78
- Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021;11(4):83. doi:10.1038/s41408-021-00474-8
- Corre J, Perrot A, Caillot D, et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood. 2021;137(9):1192-1195. doi:10.1182/blood.2020008346
- Walker BA, Mavrommatis K, Wardell CP, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159-170. doi: 10.1038/s41375-018-0196-8
- Szalat R, Avet-Loiseau H, Munshi NC. Gene expression profiles in myeloma: ready for the real world? Clin Cancer Res. 2016;22(22):5434-5442. doi:10.1158/1078-0432.CCR-16-0867
- Munshi PN, Vesole D, Jurczyszyn A, et al. Age no bar: a CIBMTR analysis of elderly patients undergoing autologous hematopoietic cell transplantation for multiple myeloma. Cancer. 2020;126(23):5077-5087. doi:10.1002/cncr.33171
- Lemieux C, Muffly LS, Rezvani A, et al. Outcomes with autologous stem cell transplant vs. non-transplant therapy in patients 70 years and older with multiple myeloma. Bone Marrow Transplant. 2021;56(2):368-375. doi:10.1038/s41409-020-01026-7
- Wildes TM, Finney JD, Fiala M, et al. High-dose therapy and autologous stem cell transplant in older adults with multiple myeloma. Bone Marrow Transplant. 2015;50(8):1075-1082. doi:10.1038/bmt.2015.106
- Sears-Smith MB, Charboneau L, Raj R, Heidel RE. Utilization of stem cell transplantation in newly diagnosed multiple myeloma patients lacking geographic proximity to a transplant center. Blood. 2021;138(suppl 1):4085. doi:10.1182/blood-2021-147895
- Yun H, Dossul T, Bernal-Mizrachi L, et al. Referral patterns and clinical outcomes for transplant-eligible lymphoma and myeloma patients evaluated at an urban county hospital. J Stem Cell Res Ther. 2016;6:328. doi:10.4172/2157-7633.1000328
- Bhatnagar V, Wu Y, Goloubeva OG, et al. Disparities in black and white patients with multiple myeloma referred for autologous hematopoietic transplantation: a single center study. Cancer. 2015;121(7):1064-1070. doi:10.1002/cncr.29160
- Schriber JR, Hari PN, Ahn KW, et al. Hispanics have the lowest stem cell transplant utilization rate for autologous hematopoietic cell transplantation for multiple myeloma in the United States: a CIBMTR report. Cancer. 2017;123(16):3141-3149. doi:10.1002/cncr.30747
- Elnair R, Holstein S. Treatment considerations for transplant-ineligible multiple myeloma. Oncology (Williston Park). 2021;35(4):170-182. doi:10.46883/ONC.2021.3504.0170
- Facon T, Kumar S, Plesner T, et al; MAIA Trial Investigators. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115. doi:10.1056/NEJMoa1817249
- Nathwani N, Kurtin SE, Lipe B, et al. Integrating touchscreen-based geriatric assessment and frailty screening for adults with multiple myeloma to drive personalized treatment decisions. JCO Oncol Pract. 2020;16(1):e92-e99. doi:10.1200/JOP.19.00208
- Wildes TM, Campagnaro E. Management of multiple myeloma in older adults: gaining ground with geriatric assessment. J Geriatr Oncol. 2017;8(1):1-7. doi:10.1016/j.jgo.2016.04.001
- Wildes TM, Tuchman SA, Klepin HD, et al. Geriatric assessment in older adults with multiple myeloma. J Am Geriatr Soc. 2019;67(5):987-991. doi:10.1111/jgs.15715
- Rosko AE, Huang Y, Benson DM, et al. Use of a comprehensive frailty assessment to predict morbidity in patients with multiple myeloma undergoing transplant. J Geriatr Oncol. 2019;10(3):479-485. doi:10.1016/j.jgo.2018.05.015
- Patel BG, Luo S, Wildes TM, Sanfilippo KM. Frailty in older adults with multiple myeloma: a study of US veterans. JCO Clin Cancer Inform. 2020;4:117-127. doi:10.1200/CCI.19.00094
- Mian HS, Wildes TM, Fiala MA. Development of a Medicare Health Outcomes Survey deficit-accumulation frailty index and its application to older patients with newly diagnosed multiple myeloma. JCO Clin Cancer Inform. 2018;2:CCI.18.00043. doi:10.1200/CCI.18.00043
- Palumbo A, Bringhen S, Mateos MV, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood. 2015;125(13):2068-2074. doi:10.1182/blood-2014-12-615187
- Engelhardt M, Dold SM, Ihorst G, et al. Geriatric assessment in multiple myeloma patients: validation of the International Myeloma Working Group (IMWG) score and comparison with other common comorbidity scores. Haematologica. 2016;101(9):1110-1119. doi:10.3324/haematol.2016.148189
- Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-M156. doi:10.1093/gerona/56.3.m146
- Engelhardt M, Domm A-S, Dold SM, et al. A concise revised Myeloma Comorbidity Index as a valid prognostic instrument in a large cohort of 801 multiple myeloma patients. Haematologica. 2017;102(5):910-921. doi:10.3324/haematol.2016.162693
- Facon T, Dimopoulos MA, Meuleman N, et al. A simplified frailty scale predicts outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma treated in the FIRST (MM-020) trial. Leukemia. 2020;34(1):224-233. doi:10.1038/s41375-019-0539-0
- Cook G, Royle K-L, Pawlyn C, et al. A clinical prediction model for outcome and therapy delivery in transplant-ineligible patients with myeloma (UK Myeloma Research Alliance Risk Profile): a development and validation study. Lancet Haematol. 2019;6(3):e154-e166. doi:10.1016/S2352-3026(18)30220-5
- Bringhen S, D’Agostino M, Paris L, et al. Lenalidomide-based induction and maintenance in elderly newly diagnosed multiple myeloma patients: updated results of the EMN01 randomized trial. Haematologica. 2020;105(7):1937-1947. doi:10.3324/haematol.2019.226407
- Facon T, Cook G, Usmani SZ, et al. Daratumumab plus lenalidomide and dexamethasone in transplant-ineligible newly diagnosed multiple myeloma: frailty subgroup analysis of MAIA. Leukemia. 2022;36(4):1066-1077. doi:10.1038/s41375-021-01488-8
- Mateos M-V, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone versus bortezomib, melphalan, and prednisone in transplant-ineligible newly diagnosed multiple myeloma: frailty subgroup analysis of ALCYONE. Clin Lymphoma Myeloma Leuk. 2021;21(11):785-798. doi:10.1016/j.clml.2021.06.005
- Stege CAM, Nasserinejad K, van der Spek E, et al. Ixazomib, daratumumab, and low-dose dexamethasone in frail patients with newly diagnosed multiple myeloma: the Hovon 143 study. J Clin Oncol. 2021;39(25):2758-2767. doi:10.1200/JCO.20.03143
- Cook G, Pawlyn C, Royle K-L, et al. FiTNEss, a UK Myeloma Research Alliance (UK-MRA) frailty-adjusted therapy study, supports the feasibility of recruiting frail newly diagnosed myeloma patients to large clinical trials. Blood. 2021;138(suppl 1):81. doi:10.1182/blood-2021-146650
- Kumar SK, Jacobus SJ, Cohen AD, et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020;21(10):1317-1330. doi:10.1016/S1470-2045(20)30452-6
- Usmani SZ, Hoering A, Ailawadhi S, et al; SWOG1211 Trial Investigators. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 2021;8(1):e45-e54. doi:10.1016/S2352-3026(20)30354-9
- Kaiser MF, Hall A, Walker K, et al. Daratumumab, cyclophosphamide, bortezomib, lenalidomide, dexamethasone (Dara-CVRd), V-augmented autologous stem cell transplant (V-ASCT) and Dara-Vrd consolidation in ultra-high risk (UHiR) newly diagnosed myeloma (NDMM) and primary plasma cell leukemia (pPCL) compared with Myeloma XI/XI+ trial treatment for UHIR MM: the UK Optimum/Muknine trial. Blood. 2021;138(suppl 1):465. doi:10.1182/blood-2021-144990
- Gay F, Musto P, Rota-Scalabrini D, et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. Lancet Oncol. 2021;22(12):1705-1720. doi:10.1016/S1470-2045(21)00535-0
- Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127(21):2569-2574. doi:10.1182/blood-2016-01-693580
- Attal M, Lauwers-Cances V, Hulin C, et al; IFM 2009 Study. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376(14):1311-1320. doi:10.1056/NEJMoa1611750
- Richardson PG, Jacobus SJ, Weller EA, et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N Engl J Med. Published online June 5, 2022. doi:10.1056/NEJMoa2204925
- Stadtmauer EA, Pasquini MC, Blackwell B, et al. Autologous transplantation, consolidation, and maintenance therapy in multiple myeloma: results of the BMT CTN 0702 Trial. J Clin Oncol. 2019;37(7):589-597. doi:10.1200/JCO.18.00685
- Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalan-prednisone, with or without bortezomib-lenalidomide-dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020;7(6):e456-e468. doi:10.1016/S2352-3026(20)30099-5
- Sonneveld P, Dimopoulos MA, Beksac M, et al. Consolidation and maintenance in newly diagnosed multiple myeloma. J Clin Oncol. 2021;39(32):3613-3622. doi:10.1200/JCO.21.01045
- Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29-38. doi:10.1016/S0140-6736(19)31240-1
- Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936-945. doi:10.1182/blood.2020005288
- Laubach JP, Kaufman JL, Sborov DW, et al. Daratumumab (DARA) plus lenalidomide, bortezomib, and dexamethasone (RVd) in patients (pts) with transplant-eligible newly diagnosed multiple myeloma (NDMM): updated analysis of GRIFFIN after 24 months of maintenance. Blood. 2021;138(suppl 1):79. doi:10.1182/blood-2021-149024
- Goldschmidt H, Mai EK, Nievergall E, et al. Addition of isatuximab to lenalidomide, bortezomib and dexamethasone as induction therapy for newly-diagnosed, transplant-eligible multiple myeloma patients: the phase III GMMG-HD7 trial. Blood. 2021;138(suppl 1):abstr 463. doi:10.1182/blood-2021-145097
- Costa LJ, Chhabra S, Medvedova E, et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. Published online December 13, 2021. J Clin Oncol. doi:10.1200/JCO.21.01935
- Landgren O, Hultcrantz M, Diamond B, et al. Safety and effectiveness of weekly carfilzomib, lenalidomide, dexamethasone, and daratumumab combination therapy for patients with newly diagnosed multiple myeloma: the MANHATTAN nonrandomized clinical trial. JAMA Oncol. 2021;7(6):862-868. doi:10.1001/jamaoncol.2021.0611
- Kesireddy M, Holstein SA. The era of lenalidomide maintenance therapy in multiple myeloma: settings for achieving best outcomes. Expert Rev Clin Pharmacol. 2022;15(1):19-31. doi:10.1080/17512433.2022.2032656
- Holstein SA, Suman VJ, Hillengass J, McCarthy PL. Future directions in maintenance therapy in multiple myeloma. J Clin Med. 2021;10(11):2261. doi:10.3390/jcm10112261
- Elnair RA, Holstein SA. Evolution of treatment paradigms in newly diagnosed multiple myeloma. Drugs. 2021;81(7):825-840. doi:10.1007/s40265-021-01514-0
- Attal M, Lauwers-Cances V, Marit G, et al; IFM Investigators. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782-1791. doi:10.1056/NEJMoa1114138
- McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770-1781. doi:10.1056/NEJMoa1114083
- Holstein SA, Jung SH, Richardson PG, et al. Updated analysis of CALGB (Alliance) 100104 assessing lenalidomide versus placebo maintenance after single autologous stem-cell transplantation for multiple myeloma: a randomised, double-blind, phase 3 trial. Lancet Haematol. 2017;4(9):e431-e442. doi:10.1016/S2352-3026(17)30140-0
- Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;371(10):895-905. doi:10.1056/NEJMoa1402888
- Jackson GH, Davies FE, Pawlyn C, et al; UK NCRI Haemato-oncology Clinical Studies Group. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019;20(1):57-73. doi:10.1016/S1470-2045(18)30687-9
- McCarthy PL, Holstein SA, Petrucci MT, et al. Lenalidomide maintenance after autologous stem-cell transplantation in newly diagnosed multiple myeloma: a meta-analysis. J Clin Oncol. 2017;35(29):3279-3289. doi:10.1200/JCO.2017.72.6679
- Benboubker L, Dimopoulos MA, Dispenzieri A, et al; FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906-917. doi:10.1056/NEJMoa1402551
- Durie BGM, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777): a randomised, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527. doi:10.1016/S0140-6736(16)31594-X
- Durie BGM, Hoering A, Sexton R, et al. Longer term follow-up of the randomized phase III trial SWOG S0777: bortezomib, lenalidomide and dexamethasone vs. lenalidomide and dexamethasone in patients (pts) with previously untreated multiple myeloma without an intent for immediate autologous stem cell transplant (ASCT). Blood Cancer J. 2020;10(5):53. doi:10.1038/s41408-020-0311-8
- O’Donnell EK, Laubach JP, Yee AJ, et al. A phase 2 study of modified lenalidomide, bortezomib and dexamethasone in transplant-ineligible multiple myeloma. Br J Haematol. 2018;182(2):222-230. doi:10.1111/bjh.15261
- Facon T, Kumar SK, Plesner T, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(11):1582-1596. doi:10.1016/S1470-2045(21)00466-6
- Facon T, Venner CP, Bahlis NJ, et al. Oral ixazomib, lenalidomide, and dexamethasone for transplant-ineligible patients with newly diagnosed multiple myeloma. Blood. 2021;137(26):3616-3628. doi:10.1182/blood.2020008787
- Usmani SZ, Schjesvold F, Oriol A, et al; KEYNOTE-185 Investigators. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol. 2019;6(9):e448-e458. doi:10.1016/S2352-3026(19)30109-7
- Facon T, Lee JH, Moreau P, et al. Carfilzomib or bortezomib with melphalan-prednisone for transplant-ineligible patients with newly diagnosed multiple myeloma. Blood. 2019;133(18):1953-1963. doi:10.1182/blood-2018-09-874396
- Mateos M-V, Cavo M, Blade J, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet. 2020;395(10218):132-141. doi:10.1016/S0140-6736(19)32956-3
- Mateos M-V, Dimopoulos MA, Cavo M, et al; ALCYONE Trial Investigators. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528. doi:10.1056/NEJMoa1714678
- Usmani SZ, Alonso Alonso A, Quach H, et al. DREAMM-9: phase I study of belantamab mafodotin plus standard of care in patients with transplant-ineligible newly diagnosed multiple myeloma. Blood. 2021;138(suppl 1):abstr 2738. doi:10.1182/blood-2021-153315
- White DJ, LeBlanc R, Baljevic M, et al. Selinexor, lenalidomide and dexamethasone (SRd) for patients with relapsed/refractory and newly diagnosed multiple myeloma. Blood. 2020;136(suppl 1):45-46. doi:10.1182/blood-2020-140141
- Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727-738. doi:10.1056/NEJMoa1903455
- van de Donk NWCJ, Popat R, Larsen J, et al. First results of iberdomide (IBER; CC-220) in combination with dexamethasone (DEX) and daratumumab (DARA) or bortezomib (BORT) in patients with relapsed/refractory multiple myeloma (RRMM). Blood. 2020;136(suppl 1):16-17. doi:10.1182/blood-2020-137743
- Richardson PG, Vangsted AJ, Ramasamy K, et al. First-in-human phase I study of the novel CELMoD agent CC-92480 combined with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2020;38(suppl 15):abstr 8500. doi:10.1200/JCO.2020.38.15_suppl.8500
- Holstein SA, Suman VJ, McCarthy PL. Should overall survival remain an endpoint for multiple myeloma trials? Curr Hematol Malig Rep. 2019;14(1):31-38. doi:10.1007/s11899-019-0495-9
- Costa LJ, Derman BA, Bal S, et al. International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials. Leukemia. 2021;35(1):18-30. doi:10.1038/s41375-020-01012-4
- Diamond BT, Rustad E, Maclachlan K, et al. Defining the undetectable: the current landscape of minimal residual disease assessment in multiple myeloma and goals for future clarity. Blood Rev. 2021;46:100732.doi:10.1016/j.blre.2020.100732
- Solimando AG, Da Vià MC, Cicco S, et al. High-risk multiple myeloma: integrated clinical and omics approach dissects the neoplastic clone and the tumor microenvironment. J Clin Med. 2019;8(7):997. doi:10.3390/jcm8070997
- Goldschmidt H, Mai EK, Bertsch U, et al. Elotuzumab in combination with lenalidomide, bortezomib, dexamethasone and autologous transplantation for newly-diagnosed multiple myeloma: results from the randomized phase III GMMG-HD6 trial. Blood. 2021;138(suppl 1):abstr 486. doi:10.1182/blood-2021-147323







: An Ongoing Problem in Cancer Treatment")






Navigating AE Management for Cellular Therapy Across Hematologic Cancers
A panel of clinical pharmacists discussed strategies for mitigating toxicities across different multiple myeloma, lymphoma, and leukemia populations.
![“[This approval] will be a quite dramatic change in our philosophy and practice in multiple myeloma," according to Joseph Mikhael, MD, MEd, FRCPC, FACP, FASCO.](/_next/image?url=https%3A%2F%2Fcdn.sanity.io%2Fimages%2F0vv8moc6%2Fcancernetwork%2F3cab3ada4c023b68c118240a512e31d72a7f931b-1200x628.png%3Ffit%3Dcrop%26auto%3Dformat&w=3840&q=75 "“[This approval] will be a quite dramatic change in our philosophy and practice in multiple myeloma,\" according to Joseph Mikhael, MD, MEd, FRCPC, FACP, FASCO.")


