Systemic Malignancies as a Cause of Unexpected Microangiopathic Hemolytic Anemia and Thrombocytopenia
This review describes the clinical features that should suggest a search for systemic malignancy as the cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia.
Tumor cells from malignancies of any type-carcinoma, sarcoma, lymphoma, leukemia-may cause systemic arteriolar and capillary obstructions. The high shear rates of blood passing through these obstructions result in fragmentation of the red cells and can cause severe anemia, described as microangiopathic hemolytic anemia (MAHA). The thrombi caused by these obstructions consume platelets and can lead to severe thrombocytopenia. MAHA (defined by fragmented red cells on the peripheral blood smear and evidence of hemolysis) and thrombocytopenia are the clinical features of syndromes described as thrombotic microangiopathies (TMAs). If a malignancy is not recognized as the cause of TMA, the diagnosis of thrombotic thrombocytopenic purpura (TTP) may be considered and plasma exchange, the essential treatment for TTP, may be initiated-a critical decision because this treatment carries a high risk of serious complications. This review describes the clinical features that should suggest a search for systemic malignancy as the cause of unexpected MAHA and thrombocytopenia. Recognition of a systemic malignancy is critical to the initiation of appropriate chemotherapy and avoidance of inappropriate use of plasma exchange treatment.
FIGURE



Brain and Lung Pathology From a Patient With a History of Breast Cancer Who Was Considered to Be in Remission
Occult malignancies as a cause of unexpected microangiopathic hemolytic anemia (MAHA) and thrombocytopenia are an uncommon but critical clinical problem. The clinical problem is emphasized and illustrated by a patient whose case we have previously reported, a 52-year-old woman who presented with sudden onset of acute abdominal pain and syncope 3 years after resection of breast carcinoma and adjuvant chemotherapy.[1] Her hematocrit was 26% with evidence of microangiopathic hemolysis (many fragmented red cells on her blood smear, increased indirect bilirubin [3.9 mg/dL] and lactate dehydrogenase [LDH; 1431 U/L] levels, negative direct Coombs test), and her platelet count was 17,000/µL. Coagulation studies, including fibrinogen level, were normal. She had been in continuous remission for the past 3 years, and computed tomographic (CT) scans showed no evidence of recurrent carcinoma. Thrombotic thrombocytopenic purpura (TTP) therefore was assumed to be the cause of this sudden illness, and plasma exchange treatment was started. The patient developed respiratory distress; spiral CT documented multiple bilateral pulmonary emboli and echocardiogram documented a dilated, akinetic right ventricle. She died 3 days after the diagnosis of TTP. I assumed that the autopsy would demonstrate multiple pulmonary emboli as the cause of her death, but I was very surprised to learn that the gross autopsy was normal. No pulmonary emboli were seen; there was no evidence of recurrent carcinoma; no cause of death was established. Therefore I assumed that she had TTP with very unusual clinical manifestations, and I assumed that the microscopic examination would demonstrate microvascular platelet thrombi in multiple organs, the characteristic pathologic abnormality of TTP. My second great surprise came several weeks later, when the pathologist called to tell me about the microscopic findings. Small intravascular clusters of metastatic carcinoma cells, consistent with breast carcinoma, were present in arterioles and capillaries of nearly every section of the lungs, brain, heart, liver, spleen, lymph nodes, and marrow. The vessels were occluded by tumor emboli and also by platelet-fibrin thrombi, which must have accumulated proximal to the tumor cells (see Figure). The platelet-fibrin thrombi were similar to the characteristic pathologic feature of TTP, however in this patient the thrombi were the result of microvascular obstruction by microscopic tumor emboli. Therefore this patient had acute and fatal malignancy-associated thrombotic microangiopathy (TMA) that was mistakenly diagnosed as TTP. The lesson I learned was that because of her atypical clinical features, we should have been more aggressive in our evaluation, which should have included a bone marrow biopsy. This might have revealed the correct diagnosis and could have enabled the patient to avoid inappropriate plasma exchange treatment.
Pathogenesis of Malignancy-Associated Microangiopathic Hemolytic Anemia (MAHA) and Thrombocytopenia
The term "microangiopathic hemolytic anemia" was used by Brain et al in their initial description of mechanical red cell destruction in 1962. They observed that "widespread intra-capillary and intra-arteriolar eosinophilic granular or amorphous thrombi" could reduce the vessel lumen to "pin-point size," causing intraluminal shearing of red cells.[2] They described the morphologic appearance of red cell fragmentation as "burr, triangular, and helmet red cells."[2] In their report describing 25 patients, the variety of conditions that caused MAHA, which almost always was accompanied by thrombocytopenia, was emphasized: 6 patients had TTP; 6 had malignant hypertension; 8 had acute renal diseases of multiple etiologies; and 5 had metastatic carcinomas (stomach, 3; lung, 1; prostate, 1).[2] The pathologic picture of thrombosis in arterioles and capillaries, which is associated with swelling of endothelial cells and the subendothelial space that causes MAHA and thrombocytopenia, is described as thrombotic microangiopathy.[3] Since the initial report of MAHA,[2] multiple other disorders have been recognized as potential causes of TMA: hemolytic-uremic syndrome (HUS), scleroderma, antiphospholipid antibody syndrome, drug toxicity, pre-eclampsia, radiation nephropathy, renal allograft rejection, hematopoietic stem cell transplantation, and human immunodeficiency virus (HIV) infection.[3,4]
In 1979, Antman et al reported on 4 patients with cancer and MAHA and reviewed 51 previously reported cases.[5] Although more than half of the 55 patients had coagulation abnormalities consistent with disseminated intravascular coagulation (DIC), Antman et al proposed that the primary cause of hemolytic anemia and thrombocytopenia is the mechanical factor of vascular obstruction by tumor cell emboli. Their conclusion was that systemic malignancies of many different types can cause both the pathologic and clinical features of TMA without abnormalities of coagulation.
Frequency of Systemic Malignancies That Can Cause MAHA and Thrombocytopenia Misdiagnosed as TTP
TABLE 1


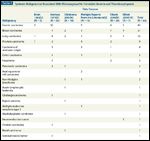
Systemic Malignancies Associated With Microangiopathic Hemolytic Anemia and Thrombocytopenia
In most patients with malignancy-associated MAHA and thrombocytopenia, the systemic malignancy is already known to be the patient’s primary problem. It is uncommon for MAHA and thrombocytopenia to be the first clinical manifestation of a systemic malignancy. It is this uncommon presentation, however, that requires awareness and appropriate evaluation by the physician. A report on the experience of the Oklahoma TTP-HUS Registry described 10 patients initially diagnosed with TTP, treated with plasma exchange, and subsequently diagnosed as having MAHA and thrombocytopenia caused by a systemic malignancy.[6] Two patients in the Registry with malignancy-associated MAHA and thrombocytopenia were not included in this report because metastatic carcinomas (one lung, one prostate) were apparent at the time that their physicians diagnosed TTP. If I had been asked, I would have advised these physicians against the use of plasma exchange, because these patients did not have the diagnostic criteria for TTP, which are MAHA and thrombocytopenia without another clinically apparent etiology.[4] During the 5 years since the preparation of their report, no additional patients in the Oklahoma Registry have had a systemic malignancy misdiagnosed as TTP. Among the 415 patients in the Oklahoma TTP-HUS Registry who were clinically diagnosed with their initial episode of TTP over a period of more than two decades, from 1989 to 2010, only the 10 patients (2.4%) described in the report were subsequently diagnosed with an initially unsuspected systemic malignancy.[6] Therefore this is an uncommon occurrence, with a frequency of less than 1 patient per year in a region with a population of approximately 2 million. This low frequency is consistent with our systematic literature review in 2006 that identified only 19 additional patients who were initially diagnosed with TTP, treated with plasma exchange, and then subsequently diagnosed as having a systemic malignancy as the cause of their MAHA and their thrombocytopenia.[6]
The Variety of Systemic Malignancies That Can Cause MAHA and Thrombocytopenia Misdiagnosed as TTP
Table 1 lists 19 different systemic malignancies that have been reported to cause MAHA and thrombocytopenia. The conclusions from these data are that MAHA and thrombocytopenia are more frequently caused by the more common malignancies, but many systemic malignancies-perhaps any systemic malignancy-can cause MAHA and thrombocytopenia.
Recognition of Systemic Malignancies as the Cause of MAHA and Thrombocytopenia
TABLE 2



Characteristics of Oklahoma TTP-HUS Registry Patients With Microangiopathic Hemolytic Anemia and Thrombocytopenia Caused by Systemic Malignancy vs Patients With TTP Associated With Severe ADAMTS13 Deficiency
Table 2 describes the clinical and laboratory features that are unusual in TTP and which suggest suspicion of systemic malignancy. These features indicate that a search for an alternative etiology for the MAHA and thrombocytopenia is appropriate.
History and physical examination
Although TTP can occur at any age in both men and women, it occurs predominantly in women of middle age. Consideration of TTP in an older man suggests that a more careful evaluation for alternative etiologies is appropriate. In any patient with a previous diagnosis of cancer, a very thorough evaluation for recurrence is necessary. This is one lesson that I learned from my experience with the patient described in this article. The history of a previous malignancy is, by itself, an appropriate indication for bone marrow biopsy. In our patient, we missed the opportunity to diagnose metastatic breast cancer when we did not perform a bone marrow biopsy. Although the prodromal symptoms of TTP can occur over a period of several weeks, the typical presentation is a more acute onset. The gradual onset of systemic symptoms over a month or more is very unusual in patients with TTP, although, as in the 52-year-old patient described in this article, MAHA and thrombocytopenia caused by an occult systemic malignancy can appear suddenly. Patients with TTP may have profound weakness and dyspnea caused by the rapid onset of severe anemia, but gradual weakness, especially accompanied by weight loss and pain, should be an indication for evaluation for an occult systemic malignancy. The principal location of the microvascular metastases is often the lung. In contrast, lung involvement is rare in patients with TTP. Therefore patients with dyspnea, cough, and a pulmonary infiltrate should be assumed to have an etiology other than TTP. In our patient described above, we did not recognize that documentation of diffuse pulmonary emboli was a clue to the presence of diffuse pulmonary microvascular metastases.
Laboratory data
The peripheral blood smear may not distinguish patients with TTP from those with a systemic malignancy; both will have fragmented red cells (schistocytes) and polychromatophilic red cells (reticulocytes) consistent with microangiopathic hemolysis, as well as decreased platelets. However, patients with marrow involvement by malignancy may also have the features of a leukoerythroblastic reaction, with premature release of immature granulocytes (myelocytes, promyelocytes, or even myeloblasts) and nucleated red cells indicating disruption of the marrow structure. Hemolysis in patients with TTP is associated with an increased level of serum LDH, but the LDH increase is often much greater in patients with systemic malignancies. The presence of DIC does not exclude TTP but it is evidence against the diagnosis of TTP. Systemic malignancies can cause TMA without the coagulation abnormalities of DIC, however.
Severe ADAMTS13 deficiency, defined as less than 10% activity,[7] is a common association with acquired TTP, and is caused by autoantibodies that inhibit ADAMTS13 activity. Severe ADAMTS13 deficiency is rarely present in patients with systemic malignancies, however. Moderate decreases of ADAMTS13 activity may occur in patients with systemic malignancies, similar to moderate ADAMTS13 deficiency in multiple other disorders associated with systemic inflammation. These moderately decreased levels of ADAMTS13 activity are not relevant to the pathogenesis of malignancy-associated TMA. Among our patients with the unexpected diagnosis of a systemic malignancy, one patient who had HIV infection and unrecognized systemic Kaposi sarcoma had severe ADAMTS13 deficiency (6% activity), but there was no evidence of TTP at autopsy. We interpreted this observation as an unusual association of severe ADAMTS13 deficiency caused by his critical illness, not as a cause of his MAHA and thrombocytopenia.[6-8] Observations such as this, together with our observations that ADAMTS13 activity assays may not always detect severely deficient activity,[7] support our practice of basing initial treatment decisions for patients suspected of having TTP only on clinical criteria.[4]
The most important diagnostic test may be a bone marrow biopsy, since marrow involvement by malignant tumor cells may be the major cause of thrombocytopenia and MAHA.[6,9,10] Bone marrow biopsies were performed in 9 of our 10 patients.[6] The only patient who did not have a bone marrow biopsy was our patient described above. The diagnosis of systemic malignancy was documented in six of these nine patients, but three bone marrow biopsies did not reveal the malignancy. In these three patients, the diagnosis of extensive bilateral lung cancer in one was established by open lung biopsy after a bronchoscopic biopsy had been normal, metastatic renal cancer in the second was diagnosed by imaging, and systemic Kaposi sarcoma in the third was undiagnosed until autopsy.[6]
Response to plasma exchange treatment may also provide diagnostic information. In patients with TTP associated with severe ADAMTS13 deficiency, a response should occur within several days. Absence of a response to plasma exchange is another clue that an alternative etiology may be present.
Additional Clinical Problems
In patients with unexpected MAHA and thrombocytopenia, the diagnostic problem is not just the distinction of TTP from a systemic malignancy. There are multiple etiologies for unexpected MAHA and thrombocytopenia other than TTP and systemic malignancies, such as systemic infections,[11] malignant hypertension,[12] severe pre-eclampsia,[13] and drug toxicity.[4] In patients with systemic malignancies who are receiving chemotherapy, the chemotherapeutic agents can cause TMA. Among the 52 patients in the Oklahoma Registry in whom TTP-HUS was attributed to drug toxicity, chemotherapeutic agents accounted for 17 cases, and the causes included mitomycin C (Mitozytrex; Mutamycin) (11 patients), gemcitabine (Gemzar) (4 patients), and BCNU (bis-chloroethylnitrosourea, carmustine [BiCNU]) and pentostatin (Nipent) (1 patient each). This is consistent with previous reports that mitomycin C[14] and gemcita-bine[15-17] are the most common agents associated with TMA. The etiology is apparently cumulative, dose-dependent renal toxicity. The course is characterized by the gradual onset of renal failure with resulting MAHA and thrombocytopenia. Acute, apparently immune-mediated TMA has been reported with gemcitabine[18] and oxaliplatin (Eloxatin),[19] however.
Management of Patients With MAHA and Thrombocytopenia Caused by Systemic Malignancy
TABLE 3



Major Complications of Plasma Exchange Treatment for TTP or HUS: Experience of the Oklahoma TTP-HUS Registry, With 249 Consecutive Patients Treated for a Diagnosis of TTP or HUS, 1996–2008
There is no benefit from plasma exchange in patients with malignancy-associated TMA. If plasma exchange treatment for TTP has already begun when a systemic malignancy is discovered, it should be stopped. In the absence of documented benefit, plasma exchange is inappropriate because of the clearly documented risks. Table 3 describes complications associated with plasma exchange treatment of 249 consecutive patients diagnosed with TTP or HUS over a period of 12 years.[20] Sixty-four (28%) patients had 72 major complications, as noted in Table 3; seven (2.8%) patients died as a result of complications of plasma exchange treatment. These data are different from those of other reports describing only rare and minor complications caused by plasma exchange.[21,22] In those reports, however, many patients had fewer procedures, performed for disorders less serious than TTP, and the patients may not have been followed throughout their entire hospital course.
The only appropriate management is chemotherapy treatment specific for the diagnosed malignancy, when this is possible. Because malignancies causing TMA are advanced, survival time is short and chemotherapy may not be effective. Among our eight patients diagnosed with a systemic malignancy while they were living, median survival after diagnosis of the malignancy was only 3 days.[6]
REFERENCE GUIDE
Therapeutic Agents
Mentioned in This Article
BCNU (bis-chloroethylnitrosourea, carmustine; BiCNU)
Gemcitabine (Gemzar)
Mitomycin C (Mitozytrex; Mutamycin)
Oxaliplatin (Eloxatin
Pentostatin (Nipent)
Brand names are listed in parentheses only if a drug is not available generically and is marketed as no more than two trademarked or registered products. More familiar alternative generic designations may also be included parenthetically.
For patients with TMA caused by chemotherapy drug toxicity, stopping the suspected drug is the only clearly appropriate management. This approach is often ineffective, however, as patients with mitomycin C and gemcitabine toxicity may have persistent severe renal failure.[14,16] Although plasma exchange has no documented benefit for patients with dose-dependent drug toxicity,[23] a trial of plasma exchange treatment is sometimes considered because of the severity of illness, its similarity to TTP, and the lack of other appropriate treatments.[16]
Conclusion
The diagnosis of TTP requires the presence of MAHA and thrombocytopenia, with or without neurologic or renal abnormalities, and without an apparent alternative etiology. Although plasma exchange is the required treatment for TTP,[4] a high probability of the diagnosis of TTP is important before this procedure is begun, because of the high risk of serious complications. Systemic malignancies can mimic the diagnostic features of TTP, but often there is a history of a malignant disorder or there are atypical clinical features that make the diagnosis of TTP less certain. When a systemic malignancy is considered as the etiology of unexpected MAHA and thrombocytopenia, it is appropriate to extend the evaluation before beginning plasma exchange treatment. For example, a bone marrow biopsy may reveal lymphoma or metastatic carcinoma. If a systemic malignancy is diagnosed, plasma exchange treatment should not be initiated. If a systemic malignancy is diagnosed after a patient has begun plasma exchange treatment for TTP, then the plasma exchange should be stopped and appropriate chemotherapy should be started.
Financial Disclosure:Dr. George serves as a consultant for Baxter, Inc., for the development of rADAMTS13 as a potential treatment for thrombotic thrombocytopenic purpura (TTP), and for Alexion, Inc., for the development of eculizumab (Soliris) as a potential treatment for atypical hemolytic-uremic syndrome (aHUS). He has no conflict with this topic or with the data presented in this article.
References:
References
1. Francis KK, Kojouri K, George JN. Occult systemic carcinoma masquerading as thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Community Oncology. 2005;2:339-43.
2. Brain MC, Dacie JV, Hourihane OB. Microangiopathic hemolytic anemia: the possible role of vascular lesions in pathogenesis. Br J Haematol. 1962;8:358-74.
3. Laszik ZG, Silva FG. Hemolytic uremic syndrome, thrombotic thrombocytopenia purpura, and other thrombotic microangiopathies and coagulopathies. In: Jennett JC, Olson JL, Schwartz MM, Silva FG, eds. Heptinstall’s Pathology of the Kidney, ed 6. Philadelphia, Lippincott Williams & Wilkins, 2007, pp 699-762.
4. George JN. How I treat patients with thrombotic thrombocytopenic purpura-2010. Blood. 2010;116:4060-9.
5. Antman KH, Skarin AT, Mayer RJ, et al. Micro-
angiopathic hemolytic anemia and cancer: a review. Medicine. 1979;58:377-84.
6. Francis KK, Kalyanam N, Terrell DR, et al. Disseminated malignancy misdiagnosed as thrombotic thrombocytopenic purpura: a report of 10 cases and a systematic review of the literature. The Oncologist. 2007;12:11-9.
7. Kremer Hovinga JA, Vesely SK, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500-11.
8. Benjamin M, Terrell DR, Vesely SK, et al. Frequency and significance of HIV infection among patients diagnosed with thrombotic thrombocytopenic purpura. Clin Infect Dis. 2009;48:1129-37.
9. Oberic L, Buffet M, Schwarzinger M, et al. Cancer awareness in atypical thrombotic microangiopathies. The Oncologist. 2009;14:769-79.
10. Elliott MA, Letendre L, Gastineau DA, et al. Cancer-associated microangiopathic hemolytic anemia with thrombocytopenia: an important diagnostic consideration. Eur J Haematol. 2010;85:43-50.
11. Booth KK, Terrell DR, Vesely SK, George JN. Systemic infections mimicking thrombotic thrombocytopenic purpura. Am J Hematol. 2011 June 14. [Epub ahead of print]
12. Egan JA, Bandarenko N, Hay SN, et al. Differentiating thrombotic microangiopathies induced by severe hypertension from anemia and thrombocytopenia seen in thrombotic thrombocytopenic purpura. J Clin Apher. 2004;19:125-9.
13. McMinn JR, George JN. Evaluation of women with clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome during pregnancy. J Clin Apher. 2001;16:202-9.
14. Lesesne JB, Rothschild N, Erickson B, et al. Cancer-associated hemolytic-uremic syndrome: analysis of 85 cases from a national registry. J Clin Oncol. 1989;7:781-9.
15. Humphreys BD, Sharman JP, Henderson JM, et al. Gemcitabine-associated thrombotic microangiopathy. Cancer. 2004;100:2664-70.
16. Izzedine H, Isnard-Bagnis C, Launey-Vacher V, et al. Gemcitabine-induced thrombotic microangiopathy: a systematic review. Nephrol Dial Transplant. 2006;21:3038-45.
17. Zupancic M, Shah PC, Shah-Khan F. Gemcitabine-associated thrombotic thrombocytopenic purpura. Lancet Oncol. 2007;8:634-41.
18. De Smet D, Jochmans K, Neyns B. Development of thrombotic thrombocytopenic purpura after a single dose of gemcitabine. Ann Hematol. 2008;87:495-6.
19. Dahabreh I, Tsoutsos G, Tseligas D, Janinis D. Hemolytic uremic syndrome following the infusion of oxaliplatin: case report. BMC Clin Pharmacol. 2006;6:5-8.
20. Nguyen L, Terrell DR, Duvall D, et al. Complications of plasma exchange in patients treated for thrombotic thrombocytopenic purpura. IV. An additional study of 43 consecutive patients, 2005-2008. Transfusion. 2009;49:392-4.
21. Shemin D, Briggs D, Greenan M. Complications of therapeutic plasma exchange: a prospective study of 1727 procedures. J Clin Apher. 2007;22:270-6.
22. Lu Q, Nedelcu E, Ziman A, et al. Standardized protocol to identify high-risk patients undergoing therapeutic apheresis procedures. J Clin Apher. 2008;23:111-15.
23. McCaleb E, Jones BS, Marques MB. Is therapeutic plasma exchange indicated for patients with gemcitabine-induced hemolytic uremic syndrome? J Clin Apher. 2009;24:209-14.
 and the QOPI Certification Program")
")







Late Hepatic Recurrence From Granulosa Cell Tumor: A Case Report
Granulosa cell tumors exhibit late recurrence and rare hepatic metastasis, emphasizing the need for lifelong surveillance in affected patients.


