CLL Therapy: The Increasing Importance of Predictive Markers
It is easy to anticipate that as the complex biology of CLL continues to unfold, new prognostic and predictive biomarkers will be recognized. The discovery of genetic mutations that are key regulators of the development, growth, and proliferation of CLL leukemic cells augurs an alluring future.
Chronic lymphocytic leukemia (CLL) is characterized by the progressive accumulation in blood, bone marrow, and lymphoid tissues of monoclonal B cells with a characteristic immunophenotype. The median age of patients at diagnosis is 72 years. Despite important progress in its therapy, CLL continues to be incurable. The disease has an extremely variable clinical course, response to therapy, and survival. While the overall median survival of patients with CLL is around 10 years, the individual prognosis ranges from a few months to a normal lifespan; this heterogeneity reflects the biological diversity of CLL.
Because of this, treatment needs to be individualized and take into account many factors, including prognostic markers.
Although a discussion of the prognosis of patients with CLL is beyond the scope of this commentary, a number of points are worthy of emphasis. First, prognostic factors should be validated, be simple, be easy to apply, and have independent prognostic value. Although there are a growing number of papers reporting “new” prognostic factors in CLL (and all types of cancer), most of these prognostic factors do not warrant being considered as such because they do not fulfill the above criteria. Second, prognostic factors should be separated into those that offer information about the natural history of the disease (eg, the likelihood of disease progression) (prognostic markers) and those that predict the response to a given therapy (predictive markers).[1-3]
FIGURE


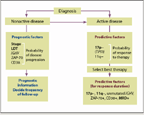
Schematic Representation of the Potential Use of Prognostic and Predictive Factors in Chronic Lymphocytic Leukemia (in the Context of Clinical Trials Only)
As with many other tumors, the focus of the study of prognostic factors in CLL is rapidly shifting from prognostic to predictive markers. Among these, genetic features are the most important (Figure).
Several years ago, with the advent of fluorescence in situ hybridization (FISH) techniques, it became clear that most patients with CLL (~ 80%) present with genetic lesions, the most common being del(13q), trisomy 12, del(11q), and del(17p).[4] All these abnormalities have important clinical correlates.[5,6] Among these genetic lesions, however, the strongest predictive marker is del(17p), which is found in 5% to 30% of patients, with the variation in frequency dependent on the time patients are studied, their prior therapy, and their refractoriness to treatment. Eighty percent of patients with del(17p) present with mutations in the TP53 gene, but TP53 mutations can also be observed in 2% to 5% of patients without del(17p); the clinical significance of these mutations is the same as that of del(17p): these patients do not respond to conventional fludarabine-based therapy.[7,8]
In this issue of ONCOLOGY, Stephens and Byrd present an updated and timely review of the challenges posed by the treatment of patients with CLL and del(17p). They discuss the bleak prognosis of these patients, which is mainly a result of their poor response to treatment; the authors also dissect current, and still unsatisfactory, treatment options, and they underline that, whenever possible, allogeneic stem-cell transplantation should be offered early in the course of the disease. They appropriately conclude by discussing the promising role of new agents such as B-cell receptor (BCR) inhibitors, cyclin-dependent kinase (CDK) inhibitors, and B-cell lymphoma 2 (BCL-2) antagonists, and they point out that some of these agents seem to be effective across all genetic groups, including del(17p).
What are the general messages communicated by the Stephens and Byrd review? One obvious point is that patients with CLL should be referred to specialized centers so that they might benefit from specialized medical care and participation in clinical trials. Regarding disease evaluation, there is some question regarding whether del(17p) (and other prognostic and predictive biomarkers) should be routinely studied. Current guidelines for CLL management do not recommend evaluating for del(17p) or other biomarkers on a routine basis.[9] However, at most academic centers, patients are offered the chance to be evaluated for genetic lesions, IGHV mutational status, ZAP-70, and CD38 expression. Performing such evaluations, within clinical studies, is reasonable provided the goal is to better understand the natural history of the disease in relationship to these markers. On the other hand, current guidelines strongly recommend investigating prognostic and predictive markers in patients entering clinical trials in order to stratify them according to their risk and in order to perform subgroup analyses.[9]
For patients who are not eligible for clinical studies, the utility of studying prognostic biomarkers is more than doubtful. A general rule in medical practice is that clinical, laboratory, and ancillary tests should only be performed if the results will influence patients’ management. Although some patients may ask (in most cases, as a result of information obtained by browsing the internet) to “get all the tests done, just in case,” there is no proof of the advantages of such an approach. It is also unclear whether “getting all the tests done” makes patients feel more reassured-or whether, on the contrary, this makes them feel more anxious.[10] Actually, most patients can be easily and accurately tracked with regular clinical examination and simple blood parameters (eg, blood cells counts, lymphocyte doubling time).
Another important and related point rightly made by Stephens and Byrd is that the detection of del(17p) (or of other biomarkers associated with a poor prognosis) is not in itself a criterion for starting therapy, since many of these patients can be followed with no therapy for long periods of time.[11-13] The concept that patients with del(17p) invariably have a poor prognosis is mainly based on data derived from subjects in trials; such patients required therapy and thus by definition had a poor prognosis. In other words, these patients are not representative of the general population of patients with CLL, a disease in which 30% to 50% of affected individuals never require therapy.
On the other hand, 20% to 40% of patients with CLL, if not more, manifest clonal evolution.[5,6] Therefore, biomarkers should be evaluated before starting treatment (a timeframe of 3 to 6 months between genetic studies and treatment initiation is generally regarded as acceptable).
What about the future? Although FISH is still the method of choice for detecting chromosomal aberrations, this technique can only identify changes specific to the probes utilized and thus underestimates the extent of existing aberrations. There is, therefore, a growing interest in applying other, more sensitive techniques to the study of genetic lesions in CLL (eg, comparative genomic hybridization, CD40- or CpG-stimulated metaphase cytogenetics, single nucleotide polymorphism arrays). Furthermore, epigenetic lesions, previously largely ignored in CLL, are increasingly being investigated.[5,6] More important, new technologies that facilitate the highly efficient sequencing of DNA have made it possible to identify new mutations (eg, NOTCH1, SF3B1, BIRC3) that do not necessarily overlap with TP53 mutations, and that seemingly are associated with a poor prognosis in patients with CLL.[14-18] These markers, however, are not yet ready for mainstream use.
Finally, it is easy to anticipate that as the complex biology of CLL continues to unfold, new prognostic and predictive biomarkers will be recognized. The discovery of genetic mutations that are key regulators of the development, growth, and proliferation of CLL leukemic cells augurs an alluring future. The time when we will be able to identify treatment-specific predictive markers and to develop targeted, more specific, and better therapies for CLL is getting closer and closer.
Financial Disclosure: The author has no significant financial interest or other relationship with the manufacturers of any products or providers of any service mentioned in this article.
References:
REFERENCES
1. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89-95.
2. Oldenhuis CN, Oosting SF, Gietma JA, de Vries EG. Prognostic versus predictive value of biomarkers in oncology. Eur J Cancer. 2008;44:946-53.
3. Mandrekar SJ, Sargent DJ. Predictive biomarker validation in practice. Lessons from real trials. Clin Trials. 2010;7:567-73.
4. Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910-16.
5. Montserrat E, Moreno C. Genetic lesions in chronic lymphocytic leukemia: clinical implications. Curr Opin Oncol. 2009;21 609-14.
6. Zenz T, Mertens D, Döhner H, Stilgenbauer S. Importance of genetics in chronic lymphocytic leukemia. Blood Rev. 2011;25:131-7.
7. Rossi D, Cerri M, Deambrogi C, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15:995-1004.
8. Gonzalez D, Martinez P, Wade R, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29:2223-9.
9. Hallek M, Cheson BD, Catovsky D, et al. International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute Working Group 1996 guidelines. Blood. 2008;111:5446-56.
10. Levin TT, LiY, Riskind K, Rai K. Depression, anxiety and quality of life in a chronic lymphocytic leukemia cohort. Gen Hosp Psychiatry. 2007;29:251-6.
11. Best OG, Gardiner AC, Davis ZA, et al. A subset of Binet stage A CLL patients with TP53 abnormalities and mutated IGHV genes have stable disease. Leukemia. 2009;23:212-4.
12. Tam CS, Shanafelt TD, Wierda WG, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114:957-64.
13. Delgado J, Espinet B, Oliveira AC, et al. Chronic lymphocytic leukaemia with 17p deletion: a retrospective analysis of prognostic factors and therapy results. Br J Haematol. 2011;157:67-74.
14. Sportoletti P, Baldoni S, Cavalli L, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br J Haematol. 2010;151:404-6.
15. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101-5.
16. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389-401.
17. Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118:6904-8.
18. Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497-506.















